Search for matches to dbCAN HMMs of CAZy carbohydrate active enzyme families using HMMER 3
This method scans protein sequences found in Genomes and Annotated Metagenome Assemblies (AMAs) using a set of Hidden Markov Models (HMMs) from the dbCAN2 CAZy collection. It uses HMMER software.
Search with dbCAN2 HMMs of CAZy families profiles collections of genes, genomes, and/or annotated metagenome assemblies for CAZy functions and optionally outputs FeatureSet collections for each of the requested gene families. It uses gene-family-derived HMMs from the dbCAN2 collection. The user can run with the entire collection, just those from a given CAZy category, or specify individual gene families with which to search. In this last mode, FeatureSet objects are produced that can be used in additional KBase phylogenomic Apps, such as Build Gene Tree. Hits by each gene family to genes in the target set are also shown in the report.
Tool and Data Sources:
HMMER v3.3.2 is installed from http://hmmer.org
The dbCAN2 HMM collection of CAZy families is downloaded from https://bcb.unl.edu/dbCAN2/download/.
Configuration:
Targets Objects: The Targets Objects may be a FeatureSet of genes, a Genome, a GenomeSet, a SpeciesTree, or an Annotated Metagenome Assembly (AMA). A HMMER search database will be automatically generated from the Targets Object.
Output FeatureSet basename: This is the basename for the objects that will contain the set of genes that are both hit and pass confidence thresholds for each model.
Other Parameters: See "Parameters" section below.
Output:
Output Object: Gene hits are captured in a FeatureSet output object. If there are additional user-defined thresholds, those are filtered out and do not appear in the object, even if they are shown in the output table. The Output object name is used as a basename to which the HMM name is prepended.
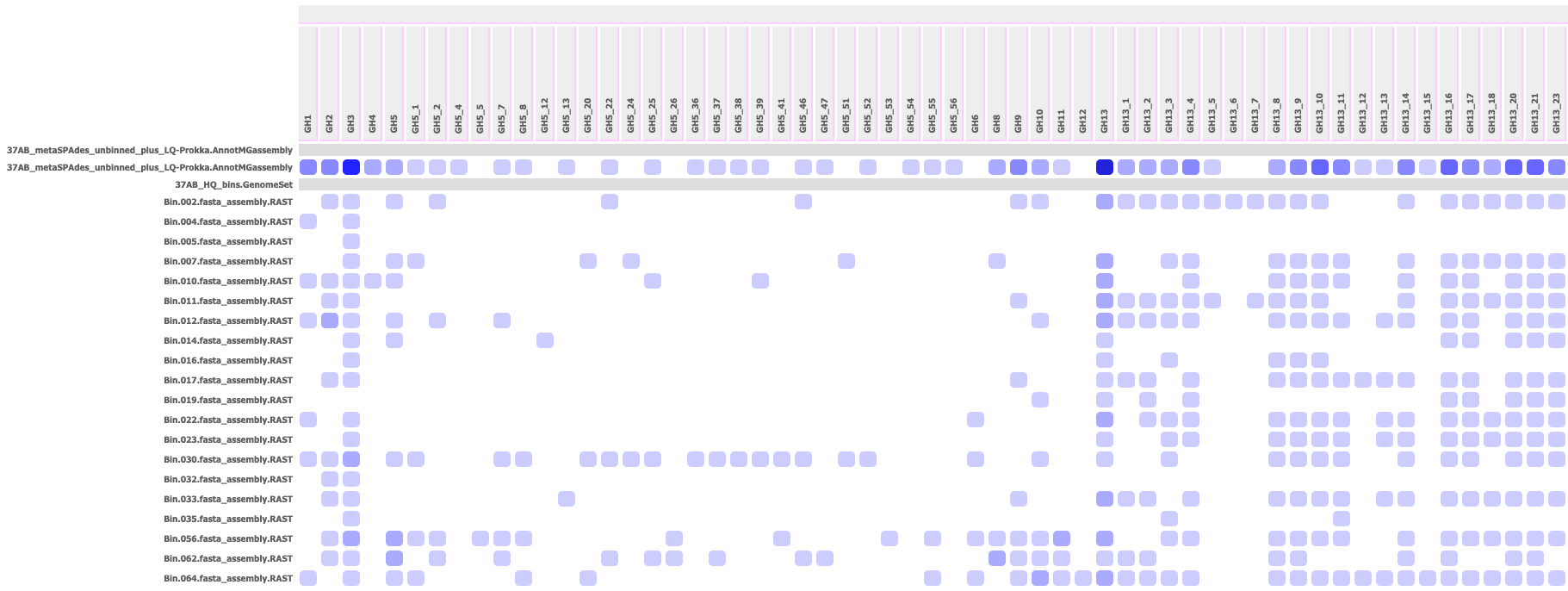
Output HTML Profile: A raw count or heatmap of the number of genes hit from each gene family (column) with each genome or annotated metagenome assembly (row). Each cell in the profile offers a roll-over of the number of hits and the gene IDs of those hits. If the input targets is a SpeciesTree, the rows are ordered by their order in a ladderized view of that tree (available from the "View Tree" App).
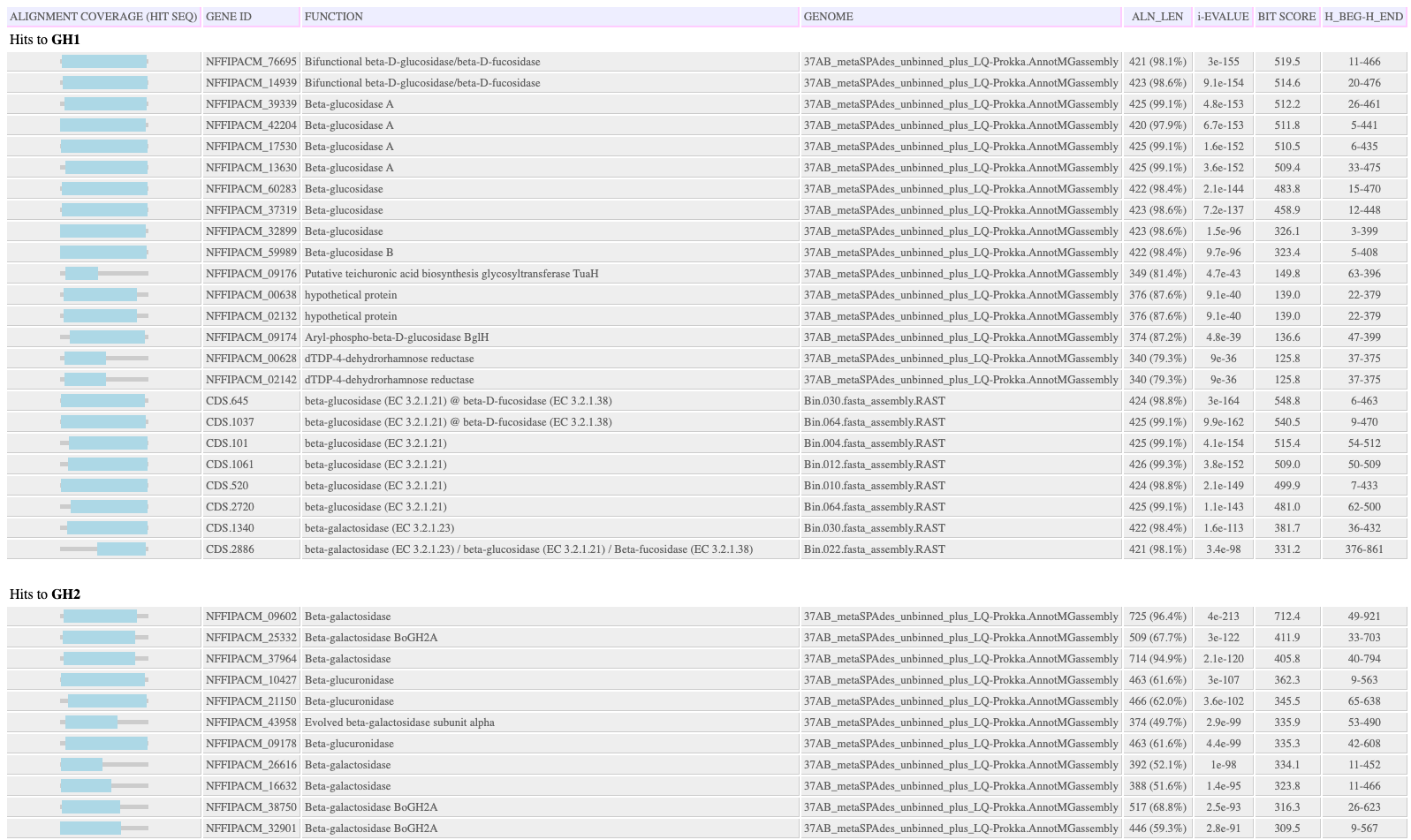
Output HTML Hit Table: The tab-delimited hit table is HTML formatted and additionally shows the region of the hit sequence (as there is no query sequence) covered by the HMMER alignment. Hits that are above the e-value threshold but below other thresholds and are not included in the FeatureSet output object are shown in gray, with the attributes that were below the threshold in red. A separate table is made for each HMM.
Downloadable files: HMMER output hit table is available for download. These are not altered from the direct output from the HMMER run. The text output is generated for each HMM.
Team members who implemented App in KBase: Dylan Chivian. For questions, please contact us.
Please cite:
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018 Jul 2;46(W1):W95-W101. doi: 10.1093/nar/gky418
- Eddy SR. Accelerated Profile HMM Searches. PLOS Computational Biology. 2011;7: e1002195. doi:10.1371/journal.pcbi.1002195
- Chivian D, Jungbluth SP, Dehal PS, Wood-Charlson EM, Canon RS, Allen BH, Clark MM, Gu T, Land ML, Price GA, Riehl WJ, Sneddon MW, Sutormin R, Zhang Q, Cottingham RW, Henry CS, Arkin AP. Metagenome-assembled genome extraction and analysis from microbiomes using KBase. Nat Protoc. 2023 Jan;18(1):208-238. doi: 10.1038/s41596-022-00747-x
Related Publications
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018 Jul 2;46(W1):W95-W101. doi: 10.1093/nar/gky418 , https://academic.oup.com/nar/article/46/W1/W95/4996582
- Eddy SR. Accelerated Profile HMM Searches. PLOS Computational Biology. 2011;7: e1002195. doi:10.1371/journal.pcbi.1002195 , https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1002195
- HMMER v3.3.2 source: , http://HMMER.ORG
- Chivian D, Jungbluth SP, Dehal PS, Wood-Charlson EM, Canon RS, Allen BH, Clark MM, Gu T, Land ML, Price GA, Riehl WJ, Sneddon MW, Sutormin R, Zhang Q, Cottingham RW, Henry CS, Arkin AP. Metagenome-assembled genome extraction and analysis from microbiomes using KBase. Nat Protoc. 2023 Jan;18(1):208-238. doi: 10.1038/s41596-022-00747-x , https://www.nature.com/articles/s41596-022-00747-x
App Specification:
https://github.com/kbaseapps/kb_hmmer/tree/6c338791492e2980534a08a1606d2d7137884759/ui/narrative/methods/HMMER_dbCAN_SearchModule Commit: 6c338791492e2980534a08a1606d2d7137884759