Allows users to compute fast whole-genome Average Nucleotide Identity (ANI) estimation.
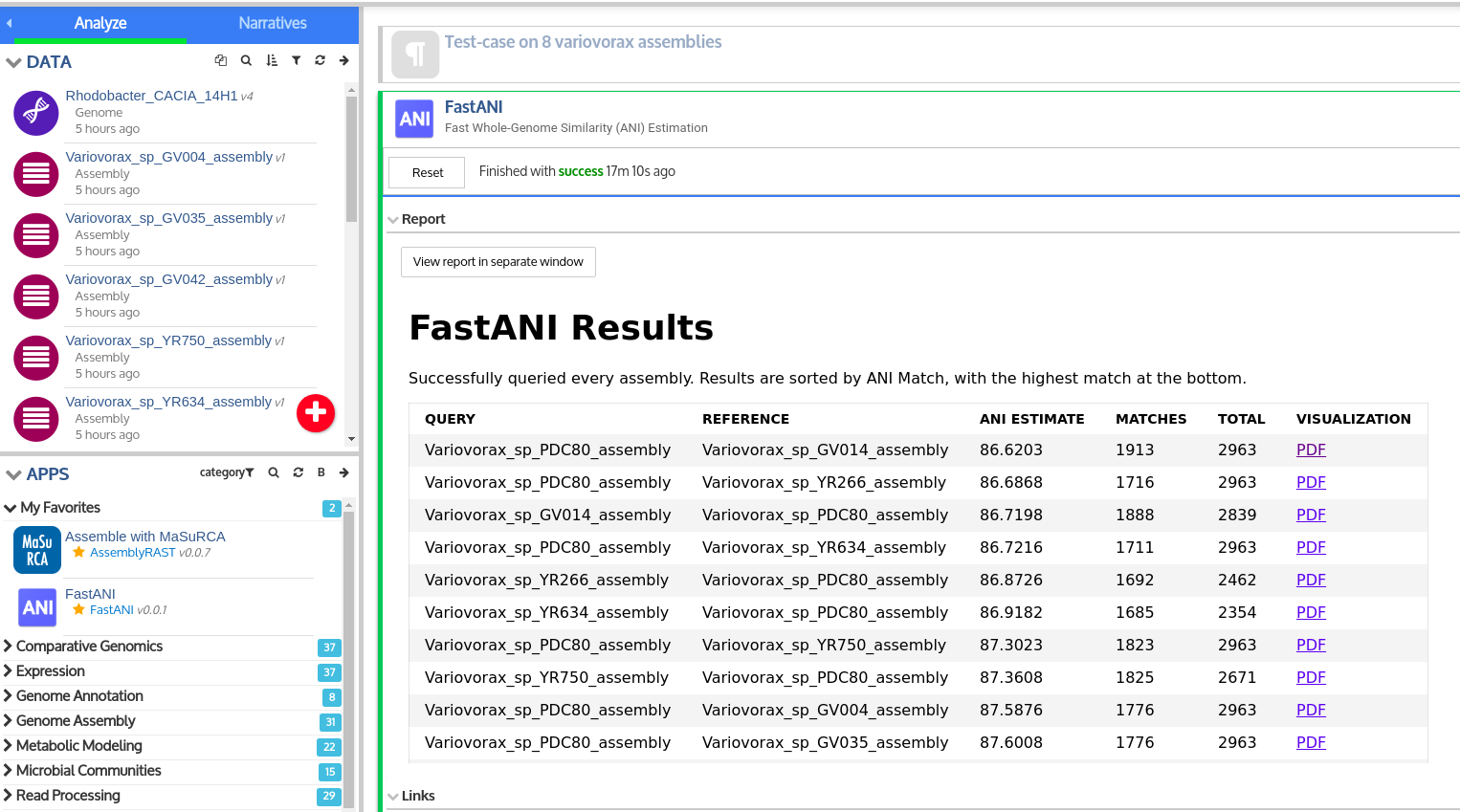
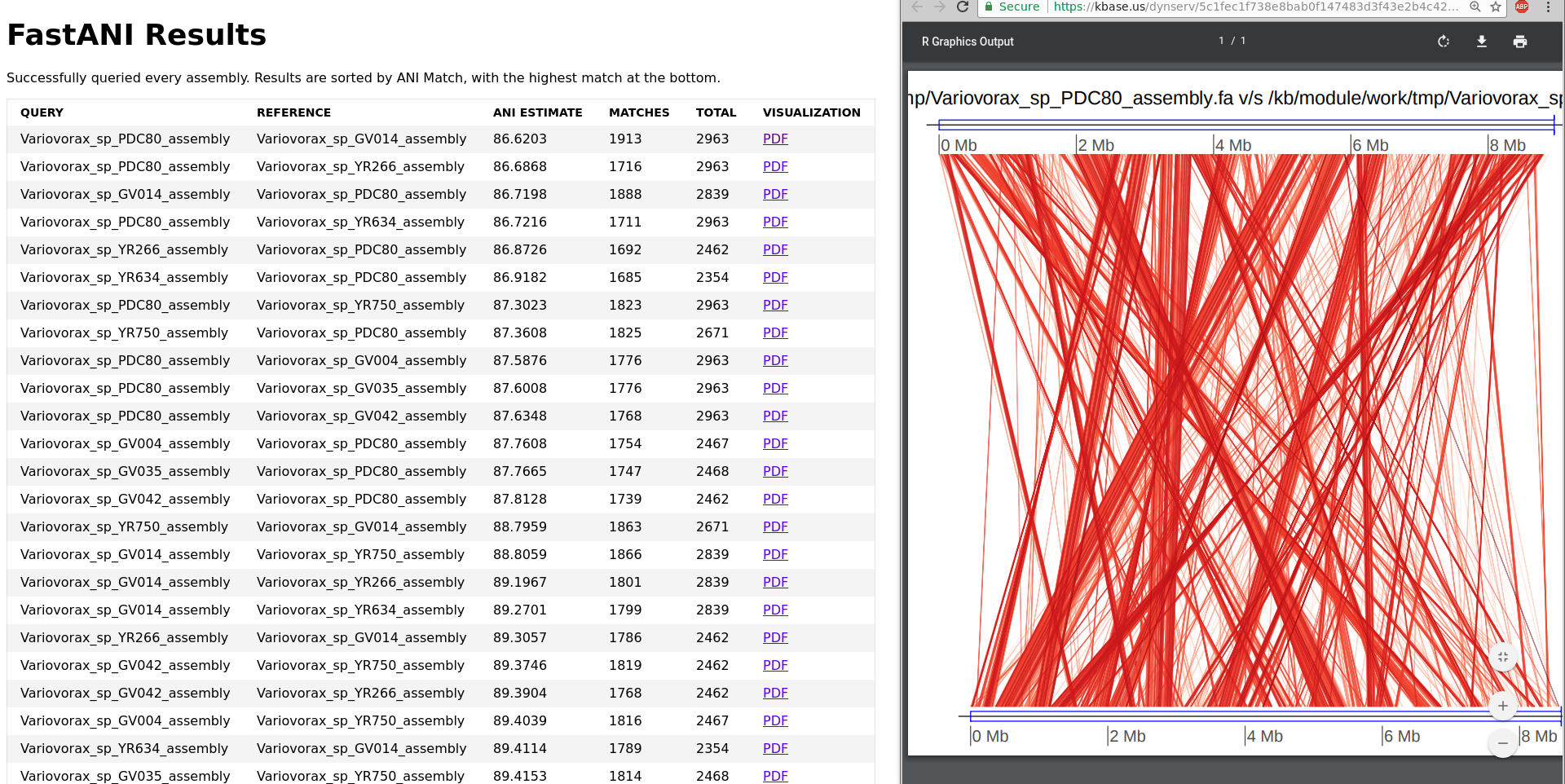
This is a KBase wrapper for FastANI, which performs fast, alignment-free computation of whole-genome Average Nucleotide Identity (ANI). ANI is defined as mean nucleotide identity of orthologous gene pairs shared between two microbial genomes. A common use of computing ANI values for assemblies would be to compare a binned assembly from a metagenome against a reference isolate to check if it is the same species as the reference isolate (>=95% ANI). In KBase, FastANI supports pairwise comparison of both complete genomes and draft genome assemblies as input. The App generates a report with the ANI values, with a PDF containing a visualization of the pairwise comparison.
FastANI produces accurate ANI estimates and is faster than alignment (e.g., BLAST)- based approaches. ANI values around 95% represent an accurate threshold for demarcating species for prokaryotes [1].
While its underlying procedure is similar to a previously described workflow [2], it avoids expensive sequence alignments and uses Mashmap as its MinHash based sequence mapping engine to compute the orthologous mappings and alignment identity estimates. Based on our experiments with complete and draft genomes, its accuracy is on par with a BLAST-based ANI solver and it achieves two to three orders of magnitude speedup. Therefore, it is useful for pairwise ANI computation of large number of genome pairs.
NOTE: No ANI output is reported for a genome pair if the ANI value is significantly below 80%. Such a case should be computed at the amino acid level.
Related Publications
- [1] Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High-throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. 2017; doi:10.1101/225342 , https://doi.org/10.1101/225342
- [2] Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57: 81 91. doi:10.1099/ijs.0.64483-0 , https://www.ncbi.nlm.nih.gov/pubmed/17220447
- FastANI module and source code: , https://github.com/ParBLiSS/FastANI
App Specification:
https://github.com/jayrbolton/kbase-fastANI/tree/5dc3c04653613cf41e3aca46d17cd4933e4356c6/ui/narrative/methods/fast_aniModule Commit: 5dc3c04653613cf41e3aca46d17cd4933e4356c6