Examine the distribution of functional gene families for organisms in a phylogenetic SpeciesTree.
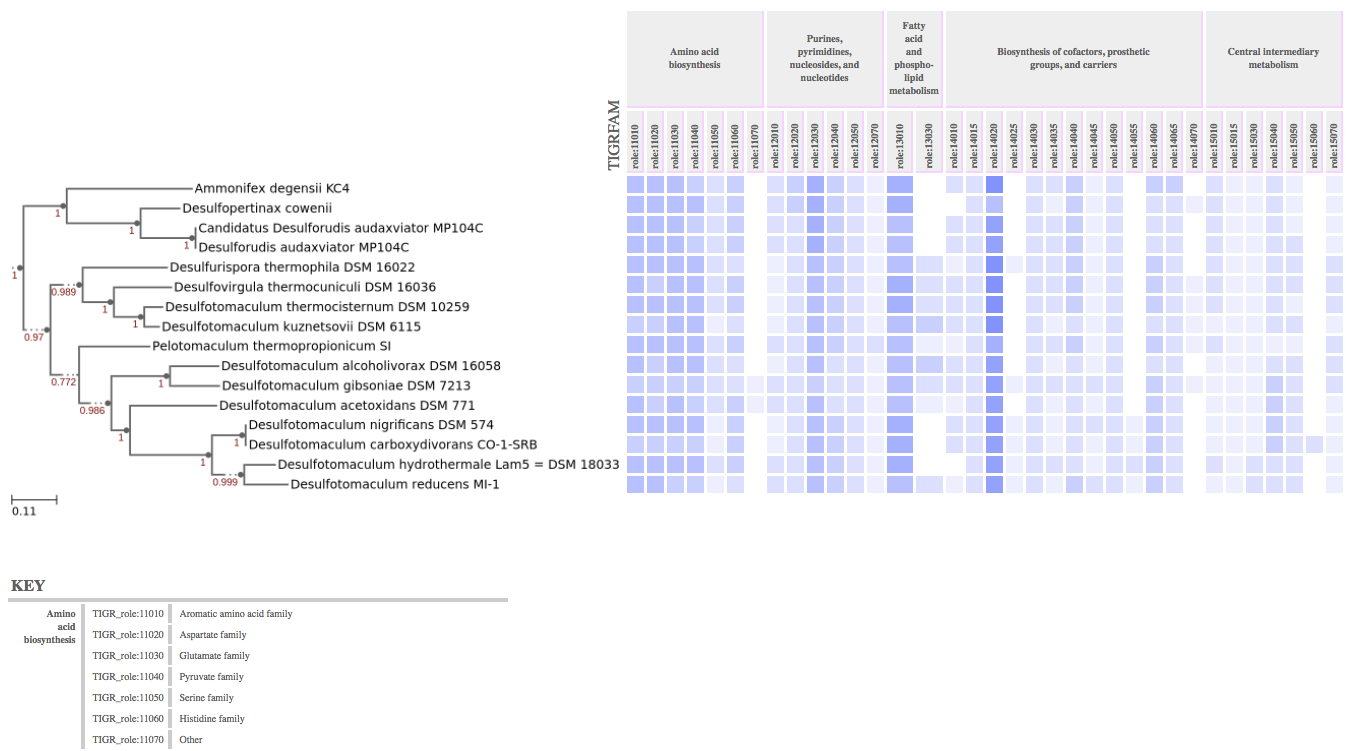
This App constructs a heatmap or numeric table showing protein domains that are present in the genomes of all the organisms contained in a phylogenetic species tree. It is recommended to use genomes annotated with RAST if you want to analyze SEED roles, but genomes annotated with Prokka or other tools are fine for other domain namespaces.
Usage
Functional assessment of an organism is often most quickly accomplished by seeing which genes carrying known functions are present in its genome. The purpose of this App is to present concise summaries of those functions, using canonical gene family assignment from COG, PFAM, TIGRFAM, and The SEED. Genomes must first be annotated using the KBase Annotate Domains in a Genome App (for each Genome separately) or the Annotate Domains in a GenomeSet App (for the Genomes on a GenomeSet or SpeciesTree) to get the COG, PFAM, and TIGRFAM annotations. For SEED annotations, the Genomes must be first be annotated by RAST using Annotate Microbial Genome App (for each Genome separately or the Annotate Multiple Microbial Genomes App (for a GenomeSet).
Functional Categories / Groups View
The tables produced by View Functional Profile can be at one of two levels of resolution. Each of the four namespaces (COG, PFAM, TIGRFAMs, The SEED) has a grouping of its families as functional "categories" or "roles". For COG, these are called "functional categories". For TIGRFAMs and The SEED, they are called "Roles". PFAM groups domain families into "Clans" which are often structurally based on SCOP. The user may request this top-level summary of the gene counts in these categories for one namespace, where the number of genes hit by the families in each category is summed.
Expanded Functional Categories / Groups View
Alternatively, the user may "drill down" into one or more categories. In "Custom" mode, the user can configure the App to report on each of the functional families with distinct counts for each domain family in each category (e.g. Alanine biosynthesis pathway genes). In this mode, there is no limitation on mixing namespaces, but some groups have many families so it's recommended not to select too many categories at first.
User-Specified Gene Families
Another Custom "drill down" option is to explicitly request custom domain families by ID (e.g. "COG0001", "PF00001", "TIGR00001", "SEED: Alanine racemase"). For each of the requested gene families, a Feature Set data object will be produced containing the genes with that annotation.
Location of Functional Annotations
Functional annotations are applied to genes either by a protein domain family (which often corresponds to a gene family, so the terms domain family and gene family are incorrectly used interchangeably henceforth). Domain Families that are functionally characterized can be used to assign function to the genes they hit with sufficient confidence. The Domain Annotation Apps produce a DomainAnnotation object which contains a link to the Genome object that it corresponds to. The View Functional Profile App will look for Domain Annotation objects in the current Narrative first. If the Genome referred to is present in a different Narrative (e.g. RefSeq genomes that haven't been copied to the current Narrative, GenomeSets and SpeciesTrees that were copied from another narrative and have not had their component Genomes updated to match the current Narrative, etc.) the "remote" narrative will also be checked for a DomainAnnotation object corresponding to the Genome.The SEED annotations are assigned using a homology-based functional assigment via the RAST annotation engine. The controlled vocabulary is based on The SEED functional subsystems. These functional assignments are not directly mapped to a collection of evolutionarily-related genes found in a gene family, so there is not an external gene family ID nor confidence and region of a hit to an HMM or multiple sequence alignment that is stored in the DomainAnnotation object such as for the other namespaces. Instead, the functional assignment is stored in the Genome object, which will have its version incremented by 1 upon RAST annotation. The View Functional Profile App looks for these functional assignments in the Genome object for SEED annotations, so may be impacted by the issue of a mismatch of the Genome object version that cannot simply be rectified by using the version from the GenomeSet or SpeciesTree. See "Troubleshooting" below if you encounter this issue.
Options
In addition to control over which functional categories and gene families are included in the analysis, the user can control confidence thresholds for annotation, summation of genes with hits to just the top hit or secondary hits, heatmap or numerical reports, percentage or raw counts, and so on as detailed below.Tree
The Species Tree containing the set of target genomes for functional profiling. Each genome will be reported as a row in the functional abundance table, ordered by the phylogenetic tree, unless it's missing sufficient annotation in the requested namespace (see "Skip Missing Genomes" and "Genes requiring NAMESPACE annot" advanced options below).Domain namespace
If using top-level functional category mode, select the annotation namespace (COG, PFAM, TIGRFAM, or The SEED) here. Otherwise, if using higher-resolution analysis mode (separate gene families), select "Use Custom Domains". Make sure you have run the DomainAnnotation App on each genome first if selecting COG, PFAM, or TIGRFAM. Make sure you have run the "Annotate Microbial Genomes with RAST" App first if selecting "SEED Roles". If you configure any Custom Domains, then Domain Namespace will be ignored.Custom domains
You may select any combination of gene families, even from different namespaces (again, as long as the Domain Annotation or RAST Annotation Apps have been run first). This may be explicit requests for domain families such as "COG0001", "PF00001", "TIGR00001", or "SEED: Alanine Racemase". If you are uncertain which domain families are most appropriate to search, it is best to use the View Functional Profile Apps iteratively, starting with nameed functional groupings and seeing which domain family IDs have hits, and then specifying them explicitly if you so choose in subsequent runs of the App. Explicity requesting a domain family by ID will produce a FeatureSet object containing the genes hit by that domain family (within the bounds for confidence and "top-hit vs. alternate hits" specified by the advanced options below).To add Custom Domains, first click on the "Enable" button, then click on the "+" button to add each Domain by ID or each functional group within each namespace.
Genome name display
The Genome may be named in the report with a user-defined combination of Object Name, Object Version, and/or Scientific Name. Choose the combination that best distinguishes each Genome for your purposes. If you have many closely related genomes with the same scientific name, you should include the Object Name to distinguish them. However, if the scientific names are all distinct and the object names are difficult to interpret, you may wish to only include the Scientific Name. It's a judgement call.Count category
Gene hits with each annotation are summed, and can either be a raw count of genes or as a percentage of genes.View table values
The functional profile table report may either contain the numbers or be represented as a heatmap (scaled to the highest value in the table). In heatmap mode, the table cells will still report their value by holding the cursor over the cell.Advanced Options
Heatmap log base
If the user wishes to view the results as a heatmap in a logarithmic scale (instead of the default linear), specify the base of the logarithm here (2 or 10 are standard, depending on the distribution of abundance values). Our suggestion for gene counts is to start with a linear report, and then if most of the signal is in just a few cells with a high count so that the heatmap bleaches the lower values and makes them difficult to discern, use a logarithmic scale to bring down the highest values and spread out the color more evenly.E-value upper limit
The DomainAnnotation App includes the confidence of the match to the domain family model as an e-value. This can be used to threshold what is considered a valid hit by the View Functional Profile App. Since smaller e-values mean greater confidence of a correct assignment, the threshold should be specified as the largest value to accept. For example, a value of 0.001 is meant to correspond to 1 erroneous assignment per 1000. That said, the actual confidence threshold is variable between domain family models, but it is beyond the capability of this particular App to offer a custom e-value threshold for each domain family. If you wish to annotate with a limited number of domain families with custom thresholds, please ask about making an App to do just that.Count only top hit or alternate hits
The e-value scores for domain hits are sorted and the user can choose to use only the most confident hit in assigning a function to a gene or use all hits (that have a confidence better than the threshold). While it may be that domain hits are non-overlapping and therefore secondary confidence hits should not be considered in conflict with the top hit, it is beyond the logic of this App at this time to include non-overlapping secondary hits as also being "top hits". The reason for this lies in the meaning of the summation of genes by "top hits" is no longer a 1:1 rule and so a clear count is lost. This is a problem with using "all hits" as well, but in this mode "double counting" is at least expected.Genes requiring COG/PFAM/TIGR/SEED annot
Annotation is an extremely imperfect endeavour. It is to be expected that only a fraction of genes in a Genome will receive annotations in any given namespace. To discern between an improperly annotated genome and one that doesn't lend itself to easy annotation (this can arise by a combination of distant phylogeny and/or novel gene families), the user may specify the percentage of genes required to be properly annotated.These thresholds only apply to namespaces configured to be part of the report, and are otherwise ignored.
In the case of The SEED annotations, the ID is the same as the functional name, and so a match of the function given for the gene is performed to verify the functions are from the same controlled vocabulary (i.e. the function "pyruvate dehydrogenase" could match even from a RefSeq annotation for a given gene, but RAST annotation is required to be run for more than coincidental matches to the controlled vocabulary). For The SEED, this percentage corresponds to genes with a functional assignment in the first place, and may also not include genes annotated as "hypothetical" (see below).
Count SEED hypothetical
The user has the option to not include genes with the annotation "hypothetical" among the annotated genes total. This only applies if the user requests functional profiling from The SEED annotations and affects the total of annotated genes for calculating the percentage with a match to The SEED controlled vocabulary (see above).Empty categories
Many namespaces have a large number of categories or domain families that will not have hits to the User's genomes. To avoid excessively large tables, the default is to not include those categories or families that have no hits to any genes. These are still included in the key below the table, but are greyed out.Skip missing genomes
It may happen that a Genome that you wish to profile does not yet have a DomainAnnotation object or been annotated by RAST. It may also be that the threshold for percentage of annotated genes has not been met, or the version doesn't match (see below). While the user will usually want to perform these annotations before running the View Functional Profile, they may instead wish to skip them from the analysis. The job log will report which genomes are missing annotations for a given namespace (or are below the required threshold for annotated genes in that namespace).Enforce genome version match
Sometimes the version of the genome object that was included in the SpeciesTree does not match the version that was used for RAST or DomainAnnotation. In the scenario that the gene coordinates are the same but the functional assignments are different, it's quite reasonable to not have to update the SpeciesTree, and instead the user may wish to allow the View Functional Profile App to ignore the version of the Genome object used for the functional annotation and instead use the version of the object from the SpeciesTree. This may be considered a "wobble" of the Genome object version.
Troubleshooting
The App fails without producing a Functional Profile Table result
The job log contains "MISSING DOMAIN ANNOTATION"
Either the indicated genomes should have the "Annotate Domains in a Genome" or the "Annotate Domains in a GenomeSet" App run to rectify the missing annotations, or the "Skip missing genomes" option should be enabled. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
The job log contains "MISSING RAST SEED ANNOTATION"
Either the indicated genomes should have the "Annotate Microbial Genomes with RAST" App run to rectify the missing annotations, or the "Skip missing genomes" option should be enabled. It may be that the genome simply doesn't have many canonical SEED functions or they are too phylogenetically remote to detect, so you can try lowering the threshold for the percentage of genes required to be match The SEED controlled vocabulary. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
The job log contains "INADEQUATE DOMAIN ANNOTATION"
Either the indicated genomes should have the "Annotate Domains in a Genome" or the "Annotate Domains in a GenomeSet" App run to rectify the missing annotations, or the "Skip missing genomes" option should be enabled. It may be that the genome simply doesn't have many canonical gene families or they are too phylogenetically remote to detect with canonical gene family models, so you can try lowering the threshold for the percentage of genes required to be annotated in that namespace. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
The job log contains "Duplicate genome display name"
Change the Genome Display option to include the Object Name, which is guaranteed to be unique (at least within a Narrative).
The App produces a Functional Profile Table that is missing Genomes
The job log contains "MISSING DOMAIN ANNOTATION"
You likely have "Skip missing genomes" enabled. Either the indicated genomes should have the "Annotate Domains in a Genome" or the "Annotate Domains in a GenomeSet" App run to rectify the missing annotations. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
The job log contains "MISSING RAST SEED ANNOTATION"
You likely have "Skip missing genomes" enabled. The indicated genomes should have the "Annotate Microbial Genomes with RAST" App run to rectify the missing annotations. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
The job log contains "INADEQUATE DOMAIN ANNOTATION"
You likely have "Skip missing genomes" enabled. Either the indicated genomes should have the "Annotate Domains in a Genome" or the "Annotate Domains in a GenomeSet" App run to rectify the missing annotations. It may be that the genome simply doesn't have many canonical gene families or they are too phylogenetically remote to detect with canonical gene family models, so you can try lowering the threshold for the percentage of genes required to be annotated in that namespace. If you are certain you already performed the annotation, you may need to "wobble" the Genome object version (see above).
I'm getting very low counts for SEED annotations
Some fraction of RefSeq annotations will match The SEED controlled vocabulary, and that may exceed the default threshold for genes required to match even though the Genome has not been RAST annotated yet. This will happen for all Genomes from the RefSeq public reference. The solution is to explicity run "Annotate Microbial Genomes with RAST" App.
I ran RAST on my genome but I'm still not seeing more SEED annotations
The SpeciesTree needs to be updated to reflect the new version of the Genome object with the update SEED annotations from RAST. Run the "Insert Set of Genomes into Species Tree" App to include the latest versions of the Genome objects after they've been annotated by RAST. Unfortunately, "wobbling" the Genome object version to match that found in the SpeciesTree is not possible in this scenario because the functional assignments are only found in the newest Genome object.Team members who developed & deployed App in KBase: Dylan Chivian, using the DomainAnnotation App by John-Marc Chandonia, Pavel Novichkov, and Roman Sutormin, and the Species Tree Builder App by Roman Sutormin and Paramvir Dehal. For questions, please contact us.
Related Publications
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nature Biotechnology. 2018;36: 566. doi: 10.1038/nbt.4163 , https://www.nature.com/articles/nbt.4163
App Specification:
https://github.com/kbaseapps/kb_phylogenomics/tree/7a7953cfae374968499ac49d7716ce9bc99b2ca0/ui/narrative/methods/view_fxn_profile_phyloModule Commit: 7a7953cfae374968499ac49d7716ce9bc99b2ca0