View a Pangenome in a phylogenetic context.
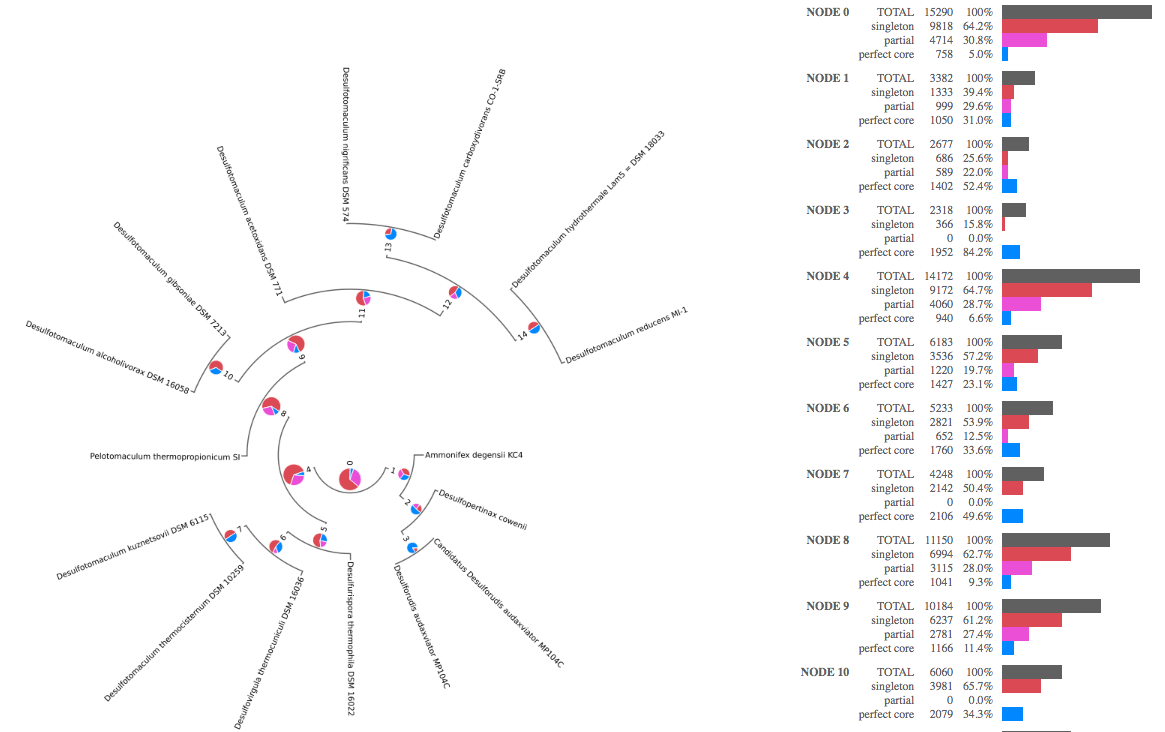
View a Pangenome in a phylogenetic context. Allows the dissection of Pangenome categories using the Species Tree so that one can determine when gene families entered or left a branch of interest.
Pangenome Categories
A Pangenome is the set of all genes found in a collection of related organisms, typically members of a species clade, which are then grouped or associated by sequence homology across species. Some methods that determine pangenomes, such as OrthoMCL, attempt to distinguish which homologous genes are true orthologs and vertically inherited from those with lineage derived paralogous expansions by duplication.
Orthologous gene sets within a pangenome are typically partitioned into three categories: Core, Singleton, and the remaining ortholog sets just considered the rest of the Pangenome (here termed the Partial Pangenome ).
- Core: The Core are those ortholog sets whose members are found in all of the collected genomes; ie each genome contains at least one gene from the ortholog set. Therefore it is most likely that the common ancestor of the species clade contained the ancestral form of the gene that each of the modern genomes inherited. Furthermore, given that the gene has been retained by all of the modern species, it is likely required, or at least sufficiently beneficial to lifestyle, to be retained. The rule used by Phylogenetic Pangenome Accumulation to determine if a homolog set should be considered Core is that a gene is found in all N genomes of the branch. When using incomplete genomes, the 100% presence rule may be too strict given that the gene may exist but not be observed. Rather than impose flexible thresholds for presence, we recommend instead that the user only include the most complete genomes possible in pangenome calculations.
- Singleton: The Pangenome calculation will find a large number of genes with no sequence homology to genes in any other genomes. These are categorized as Singletons. The rule is presence in one and only one genome. They may represent generation of novel functions, horizontal transfer from distal lineages not included in the Pangenome calculation, or missing proximal lineages that perhaps should have been included in the Pangenome calculation. If the set of genomes in the Pangenome calculation contains a broad phylogenetic sampling of the clade of interest, then the last candidate hypothesis is less likely.
- Partial Pangenome: In between Core and Singleton are those ortholog clusters present in more than one genome and fewer than all. If using incomplete genomes, these sets may contain ortholog clusters that would otherwise be calculated as core functions, so exercise caution when using incomplete genomes (absence of evidence is not evidence of absence!).
Configuration
Pangenome: The pre-calculated pangenome object (containing the orthology relationships between the genes) to use for the visualization. Only one Pangenome object can be used.
Species Tree: The phylogenetic pangenome accumulation is determined within the context of the Species Tree. Unless you select the skip missing genomes option, every Genome in the Species Tree must be contained within the Pangenome object (but not every Genome in the Pangenome object needs to be in the Species Tree). The same Narrative must be used for the Genomes found in the Pangenome and the Species Tree. Additionally, the user may choose to require that the versions of the Genome objects that were included in the Species Tree and the Pangenome must match with the Enforce Genome version match option. Only one species Tree can be used at a time.
Save Pangenome feature sets: Feature Sets containing Core, Singleton, and Partial Pangenome (see above) gene members from the ortholog clusters are created if this option is set. These categories are calculated and generated for each node in the Species Tree, using the collection of genomes for which that node represents the ancestor (i.e. the leaves of that branch).
Team members who developed & deployed algorithm in KBase: Dylan Chivian. Species Tree Builder by Roman Sutormin and Paramvir Dehal. For questions, please contact us.
Related Publications
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003;13: 2178 2189. doi:10.1101/gr.1224503 , https://genome.cshlp.org/content/13/9/2178
App Specification:
https://github.com/kbaseapps/kb_phylogenomics/tree/7a7953cfae374968499ac49d7716ce9bc99b2ca0/ui/narrative/methods/view_pan_phyloModule Commit: 7a7953cfae374968499ac49d7716ce9bc99b2ca0