Generated November 10, 2022

In this Narrative workflow, we demonstrate the KBase discovery pipeline in identifying potential gene candidates on a novel Pyridine degradation pathway in Micrococcus luteus. Here (i) we use cheminformatics analysis to propose new biochemistry then, (ii) we use metabolic modeling and omics data to identify potential gene candidates, (iii)we then query the PDB to fetch metadata/annotations for experimentally resolved structures corresponding to the gene candidates. For these selected structures, later in this workshop, PDB team will demonstrate (iv) deriving co-crystallized structures with the substrates of interest that bolster the confidence of the identified gene candidates on this novel degradation pathway. Finally, the gene candidates can be experimentally verified.
Next, we follow another interesting example in the Arabidopsis riboflavin pathway on this narrative workflow that shows the value of applying computational tools in KBase and PDB to address important scientific questions.</p>
Here we annotate the Mluteus genome using three annotation pipelines which derive functional annoations for each gene in the genome. We annotate with three seperate algorithms which increase the chances of assigning functions for maximum number of genes in the genome
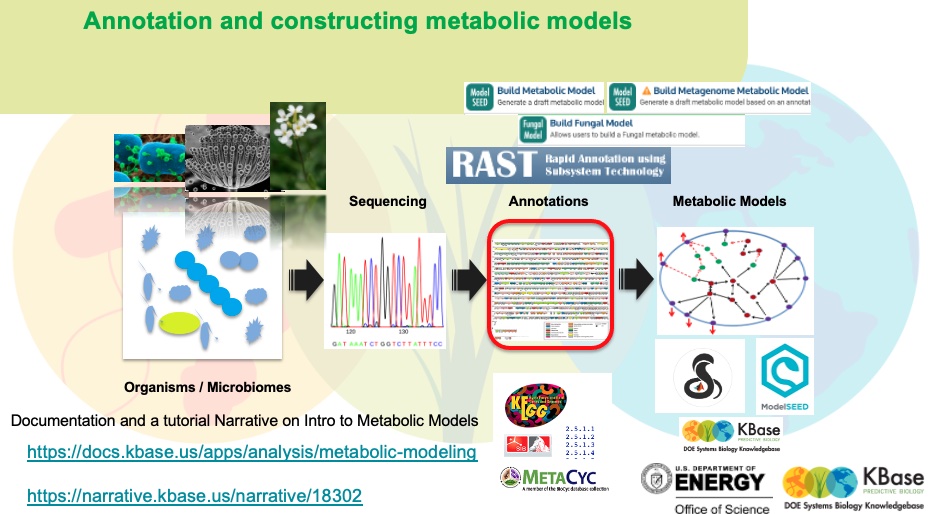
Below, the M.luteus genome is shown in a genome viewer. This viewer provides a concise, text-based overview of the genome as well as its contigs and genes.
In the Contigs and Features tabs, each entry is clickable, opening either a browser for the contig or another tab with expanded information about the gene. You can sort these entries by clicking on a column header to sort by that field (e.g., Length). Clicking the same column header again will reverse the sort order.
The M.luteus genome has two contigs. Click on one to see neighboring genes and potential operons in this species.
To further explore this genome, click on "Browse Features" tab, where you can search for gene annotations/functions by name (e.g; pyruvate synthase, EC numbers etc.), extract DNA or protein sequences, explore the neigboring genes/gene clusters


| Created Object Name | Type | Description |
|---|---|---|
| Mluteus_ATCC_49442_RAST_PROKKA | Genome | Annotated genome |

| Created Object Name | Type | Description |
|---|---|---|
| DraftModel_Mluteus | FBAModel | FBAModel-14 DraftModel_Mluteus |
| DraftModel_Mluteus.gf.1 | FBA | FBA-13 DraftModel_Mluteus.gf.1 |
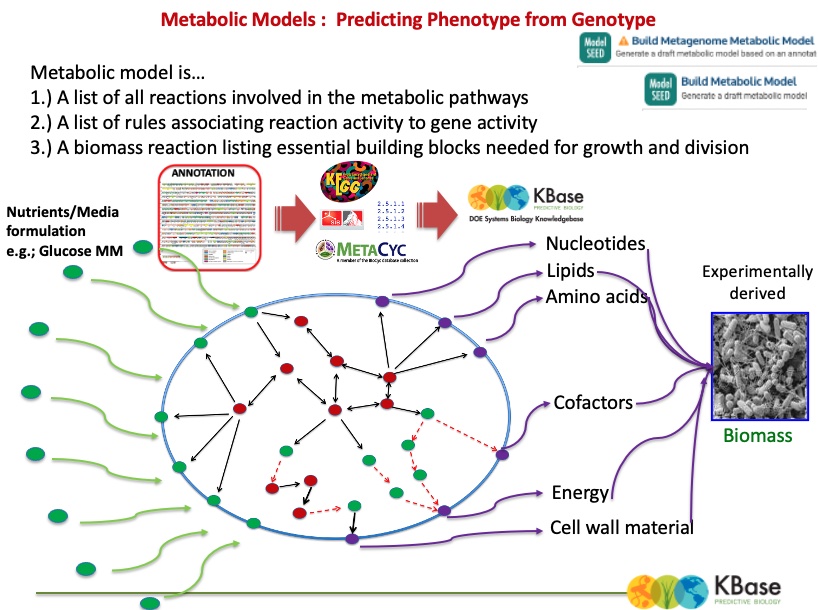
The output (above) of the Build Metabolic Model app shows information about the resulting gapfilled model. (Note that although the object type is “FBA Model,” we have not actually performed a flux balance analysis yet.)
There are eight tabs for browsing the data in the model: Overview, Reactions, Compounds, Genes, Compartments, Biomass, Gapfilling, and Pathways.
To generate some potential utilization routes for pyridine, we use Pickaxe app. This tool uses a set of general reaction rules which are curated from known biochemistry as the figure below demonstrates. These reactions can be applied to novel substrates like pyridine to propose new chemical transformations.
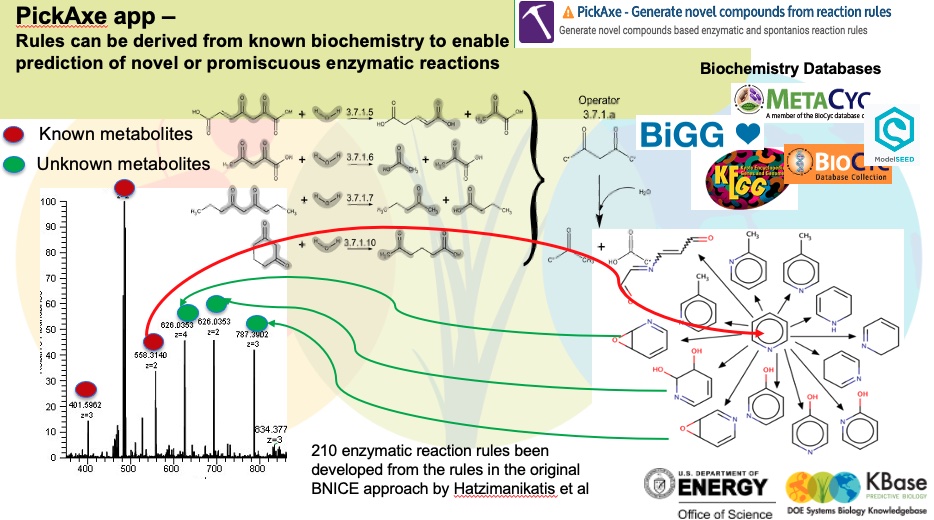

| enzymatic |
| Created Object Name | Type | Description |
|---|---|---|
| PyridineNovelReactions | FBAModel | FBAModel-14 PyridineNovelReactions |
You can construct any custom media with the Edit Media app. Here we use an existing media formulation (e.g; Glucose Minimal Media) that we can copy from our reference media and remove Glucose,the sole carbon source from the media creating a base media base media that have all necessary salts, Oxygen, Nitrogen, Sulfur, Phosphate except for the carbon source. We discuss having Pyridine as the carbon source in the following step of Gapfill Metabolic Model step (Filling knowledge gaps in the metabolic network - Gapfilling Metabolic Model).

| 489/6/5/compounds/id/cpd00027 |
| Created Object Name | Type | Description |
|---|---|---|
| BaseMedia | Media | Media-4 BaseMedia |
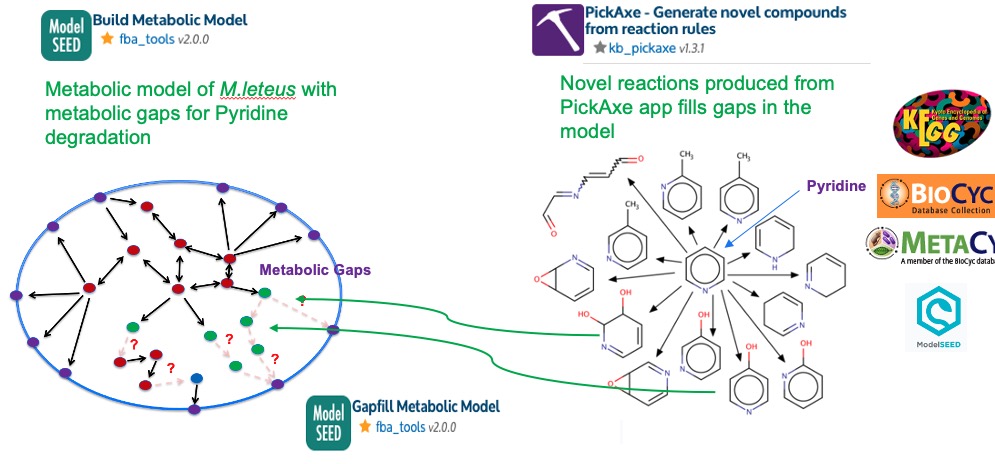
Typically, draft metabolic models tend to have metabolic gaps due to missing or incomplete annotations. In this workflow, the metaboic gap that we are interested is the Pyridine degredation as the pathway is not chracterized. We have used the PickAxe app above to generate potential noval reactions and pathways for pyridine degration. Now, in next step we use the PickAxe output (Selected under Source Gapfill Model) to fill the pyridine degredation gap in the M.luteus metabolic model.
As for the Media, we use a base media that have all necessary salts, Oxygen, Nitrogen, Sulfur, Phosphate - (Selected under Media) and the sole carbon source Pyridine will be selected under Source model media supplement option.

| bio1 |
| cpd00556_c0 |
| Created Object Name | Type | Description |
|---|---|---|
| DraftModel_MLuteus.pyridine.gf | FBAModel | FBAModel-14 DraftModel_MLuteus.pyridine.gf |
| DraftModel_MLuteus.pyridine.gf.gf.2 | FBA | FBA-13 DraftModel_MLuteus.pyridine.gf.gf.2 |
There are eight tabs for browsing the data (above) in the model: Overview, Reactions, Compounds, Genes, Compartments, Biomass, Gapfilling, and Pathways.
In order to simulate metabolic moodels (to run FBA), we need a media formulation. In this work flow we use the custom media formulation Pyridine Minimal media. You can construct any custom media with the app Edit Media. Here we use an existing media formulation (e.g; Glucose Minimal Media) that we can copy from our reference media and replace with Pyridine creating Pyridine minimal media.

| 489/6/5/compounds/id/cpd00027 |
| Created Object Name | Type | Description |
|---|---|---|
| PyridineMinimalMedia | Media | Media-4 PyridineMinimalMedia |
Now we are going to run flux balance analysis on one of the bins (genome-scale model), which will simulate how this would grow on Pyridine-minimal media.
What is FBA - https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3108565/
Flux Variability Analysis (FVA) - https://www.ncbi.nlm.nih.gov/pubmed/14642354
Reaction classifiers are assigned when using Flux Variability Analysis, FVA. In FVA, the global objective (biomass) is fixed at its optimal value, then each reaction, iteratively, is optimized independently to find both the maximal and minimal value that is possible given that the global objective must still be reached. FVA analysis composed into four categories in the following FBA. (See the column "class" in the FBA output data)

| bio1 |
| Created Object Name | Type | Description |
|---|---|---|
| FBA_Mlutues_Pyridine_Degradation | FBA | FBA-13 FBA_Mlutues_Pyridine_Degradation |
When the FBA analysis finishes, information on the flux distribution is displayed in a table with six tabs: Overview, Reaction fluxes, Exchange fluxes, Genes, Biomass, and Pathways (see above).
Genes — This tab displays the gene knockout information, if any.
Biomass — Biomass composition of the model is displayed. Typically, biomass is represented in the model as an equation where biomass compounds and ATP would make 1 gram of biomass. After clicking on the Biomass tab, the coefficients of each biomass component are listed in the Coefficient column. Negative coefficients represent the compounds on the left side of the biomass equation, and positive coefficients represent the compounds on the right side of the equation.
Now we have demonstrated a potential novel pathway for pyridine degredation, next, we can work on identifying the potential gene candidates. From the gapfilling and FBA steps, we can see the novel pyridine degradation reactions are associated with partial EC number 1.14.13 -. In our genome, there are about 30 genes are assigned with the first three digits of the EC number 1.14.13. We use the (i) differential expression data to filter out highly expressed genes (green) and (ii) the gene clustering data. Differential Expression analysis Narrative can be found here.

While the highly expressed genes with EC 1.14.13 narrow down the list of gene candidates, the gene clustering data/neighboring genes in the same operon provide valuable insights on key enzymatic steps of the degradation pathway. By surveying the gene cluster/neibouring genes with the MLuteus_masurca_RAST.CDS.3484 against PDB structural evidence, we can find the MLuteus_masurca_RAST.CDS.3483 gene, a phenylacetate dehydrogenase (paaZ) linked to a literature explaining the key step of ring opening enzyme on phenylacetate, a substrate that is chemically similar to pyridine.
Here we can query experimentally resolved structures that are corresponding to the potential gene candidates
Tune in for:
Query and learn from co-crystalized structures with the docking of the substrate
Align experimental and computational structures to aid binding site identification and functional characterization

| MTTTATAEAAVNTVETVPSFVQDSWWTPDAGSAASAVPVRDASTGEVLAKVSADGLDLAAVVEYGRTTGQAELGKLTFHQRALKLKELAQYLNARREHFYTFSAQTGATKVDSMIDIDGGIGVLFTFGSKGRRELPNSQVVVDGPMEVLSKDGSFAGEHIYTRIPGVAVQINAFNFPVWGMLEKLAPAFIAGVPTIVKPATPTGYVAAAVVKAIIESNILPAGSLQLISGSVRGLLDVLDYRDLVAFTGSASTALTLKSHRNVVEGGVRFTSETDSLNAAILGTDAVEGTPEFDAFIKSVVTEMTVKAGQKCTSIRRAIVPEGLVPAVIAAVGKRIQERVVLGDPRAEGVTMGALASVEQLADVRAAVQSMIDAGGELAYGTLDSPSVTAADGTTGVVAEGAFMSPVVLGWNDPEAEAIHSLEAFGPVASVIGYKDLPDAVRLAARGGGSLVATVCTNDPAVARELVTGIAAHHGRVLMLNREDARSSTGHGSPVPHLVHGGPGRAGGGEELGGIRSVMHHMQRTAIQGSPNMLTAVTGVWHTGADRNFTADTEGTHPFRKSLSTLHIGDAIRSELREVTLEEITKFANSTGDTFYAHTNQEAAEANPFFPGIVAHGYLLLAWGAGLFVEPAPGPVLANYGLENLRFITPVAAGDSIRVTLTAKKITPRETDEYGEVAWDAVLTNQKDEIVATYDVLTLVEKG |