Generated April 8, 2022
Nasim Maghboli Balasjin, James S. Maki, Michael R. Schläppi and Christopher W. Marshall
Marquette University, Biological Sciences Department, Milwaukee, Wisconsin, USA
Asian rice (Oryza sativa L.) is one of the most important crops because it is a staple food for almost half of the world’s population. O. sativa has two subspecies, JAPONICA and INDICA that differ by morphological, physiological and genetic characteristics. To have production of O. sativa keep pace with a growing world population, it is anticipated that the use of fertilizers will also need to increase. This increased fertilizer use may cause environmental damage through runoff impacts, but an alternative strategy to increase crop yield is the use of plant growth promoting bacteria. Thousands of microbial species can exist in association with plant roots and shoots, and some are critical to the plant’s survival. We isolated 140 bacteria from O. sativa and investigated whether JAPONICA and INDICA rice subspecies were positively influenced by these isolates. The bacterial isolates were screened for their ability to solubilize phosphate, a known plant growth promoting characteristic, and 25 isolates were selected for further analysis. These 25 phosphate solubilizing isolates were also able to produce other potentially growth-promoting factors including lipases, cellulases, proteases, siderophores, indoleacetic acid, gibberellic acid and 1-aminocyclopropane-1-carboxylic acid deaminase. Five of the most promising bacterial isolates were chosen for whole genome sequencing. Four of these bacteria, isolates related to Pseudomonas mosselii, Microvirga sp., Paenibacillus rigui and Paenibacillus graminis, improved root and shoot growth, root to shoot ratio, and increased root dry weights of JAPONICA plants but had no effect on growth and development of INDICA plants. This indicates that while bacteria have several known plant growth promoting functions, their effects on growth parameters can be plant subspecies dependent and suggest close relationships between plants and their microbial partners.

| Created Object Name | Type | Description |
|---|---|---|
| 163_S67_R1_001.fastq_reads | PairedEndLibrary | Imported Reads |


| 163_S67_R1_001.fastq_reads |
| Created Object Name | Type | Description |
|---|---|---|
| SPAdes163.contigs | Assembly | Assembled contigs |


| Created Object Name | Type | Description |
|---|---|---|
| 163_genome_annotate | Genome | Annotated genome |
The RAST algorithm was applied to annotating a genome sequence comprised of 89 contigs containing 7304039 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 7141 new features were called, of which 285 are non-coding. Output genome has the following feature types: Coding gene 6856 Non-coding crispr_array 3 Non-coding crispr_repeat 68 Non-coding crispr_spacer 65 Non-coding repeat 104 Non-coding rna 45 Overall, the genes have 2526 distinct functions. The genes include 2793 genes with a SEED annotation ontology across 1263 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions.
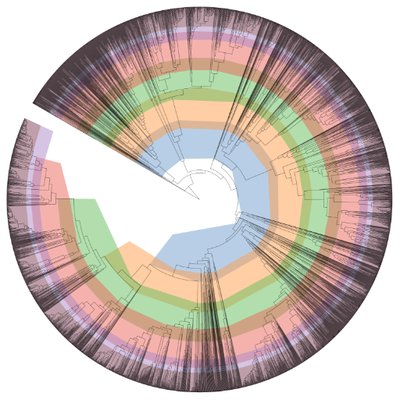

| 163_genome_annotate |

| 163_genome_annotate |
| Paenibacillus_sp._UNC451MF__GCF_000686845.1_ |
| Paenibacillus_ginsengihumi_DSM_21568__GCF_000380965.1_ |
| Paenibacillus_chitinolyticus_NBRC_15660__GCF_000739915.1_ |

| Created Object Name | Type | Description |
|---|---|---|
| 163_Nasim | Genome | Annotated genome |

| Created Object Name | Type | Description |
|---|---|---|
| 163_Nasim | DomainAnnotation | Domain Annotations |
| Created Object Name | Type | Description |
|---|---|---|
| 172_S70_R1_001.fastq_reads | PairedEndLibrary | Imported Reads |
| 172_S70_R1_001.fastq_reads |
| Created Object Name | Type | Description |
|---|---|---|
| 172_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| 172_genome_annotate | Genome | Annotated genome |
The RAST algorithm was applied to annotating a genome sequence comprised of 104 contigs containing 7161705 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 7084 new features were called, of which 122 are non-coding. Output genome has the following feature types: Coding gene 6962 Non-coding repeat 63 Non-coding rna 59 Overall, the genes have 2715 distinct functions. The genes include 2614 genes with a SEED annotation ontology across 1255 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions.
| 172_genome_annotate |

| Created Object Name | Type | Description |
|---|---|---|
| 170_S69_R1_001.fastq_reads | PairedEndLibrary | Imported Reads |
| 170_S69_R1_001.fastq_reads |
| Created Object Name | Type | Description |
|---|---|---|
| 170_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| 170_genome_annotate | Genome | Annotated genome |
The RAST algorithm was applied to annotating a genome sequence comprised of 46 contigs containing 4587927 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4652 new features were called, of which 94 are non-coding. Output genome has the following feature types: Coding gene 4558 Non-coding repeat 36 Non-coding rna 58 Overall, the genes have 2385 distinct functions. The genes include 2085 genes with a SEED annotation ontology across 1224 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions.
| 170_genome_annotate |
| 170_genome_annotate |
| Microvirga_vignae |
| Microvirga_flocculans_ATCC_BAA-817 |
| Microvirga_guangxiensis |
| Created Object Name | Type | Description |
|---|---|---|
| 167_S68_R1_001.fastq_reads | PairedEndLibrary | Imported Reads |
| 167_S68_R1_001.fastq_reads |
| Created Object Name | Type | Description |
|---|---|---|
| 167_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| 167_genome_annotate | Genome | Annotated genome |
The RAST algorithm was applied to annotating a genome sequence comprised of 153 contigs containing 6449936 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 6876 new features were called, of which 117 are non-coding. Output genome has the following feature types: Coding gene 6759 Non-coding crispr_array 1 Non-coding crispr_repeat 4 Non-coding crispr_spacer 3 Non-coding repeat 58 Non-coding rna 51 Overall, the genes have 2692 distinct functions. The genes include 3133 genes with a SEED annotation ontology across 1333 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions.
| 167_genome_annotate |
| 167_genome_annotate |
| Brevibacillus_brevis_NBRC_100599 |
| Brevibacillus_choshinensis |
| Brevibacillus_formosus |
| Created Object Name | Type | Description |
|---|---|---|
| 132_S66_001.fastq_reads | PairedEndLibrary | Imported Reads |
| 132_S66_001.fastq_reads |
| Created Object Name | Type | Description |
|---|---|---|
| 132_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| 132_genome_annotate | Genome | Annotated genome |
The RAST algorithm was applied to annotating a genome sequence comprised of 133 contigs containing 5782008 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 5448 new features were called, of which 143 are non-coding. Output genome has the following feature types: Coding gene 5305 Non-coding repeat 75 Non-coding rna 68 Overall, the genes have 3592 distinct functions. The genes include 2084 genes with a SEED annotation ontology across 1560 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions.
| 132_genome_annotate |