Generated August 14, 2020
 Delftia is a genus of bacteria with a bunch of cool features! The best studied species, Delftia acidovorans can produce gold nanoparticles from gold ions in solution. This bacteria has been found living in biofolms with Cupriavidis metallidurans on gold nuggets(1). It is also found in soil, in sinks and in rhizospheres of different plants where it promotes their growth(3).
Delftia is a genus of bacteria with a bunch of cool features! The best studied species, Delftia acidovorans can produce gold nanoparticles from gold ions in solution. This bacteria has been found living in biofolms with Cupriavidis metallidurans on gold nuggets(1). It is also found in soil, in sinks and in rhizospheres of different plants where it promotes their growth(3).
Delftia acidovorans forms gold nuggets by producing a short nonribosomal peptide called delftibactin (1). The 16 genes responsible for the production of delftibactin are called the del cluster (delA-delP) (1). These genes were originally discovered in Delftia acidovorans SPH-1, but our research shows that the del cluster appears to be present across the genus (1).
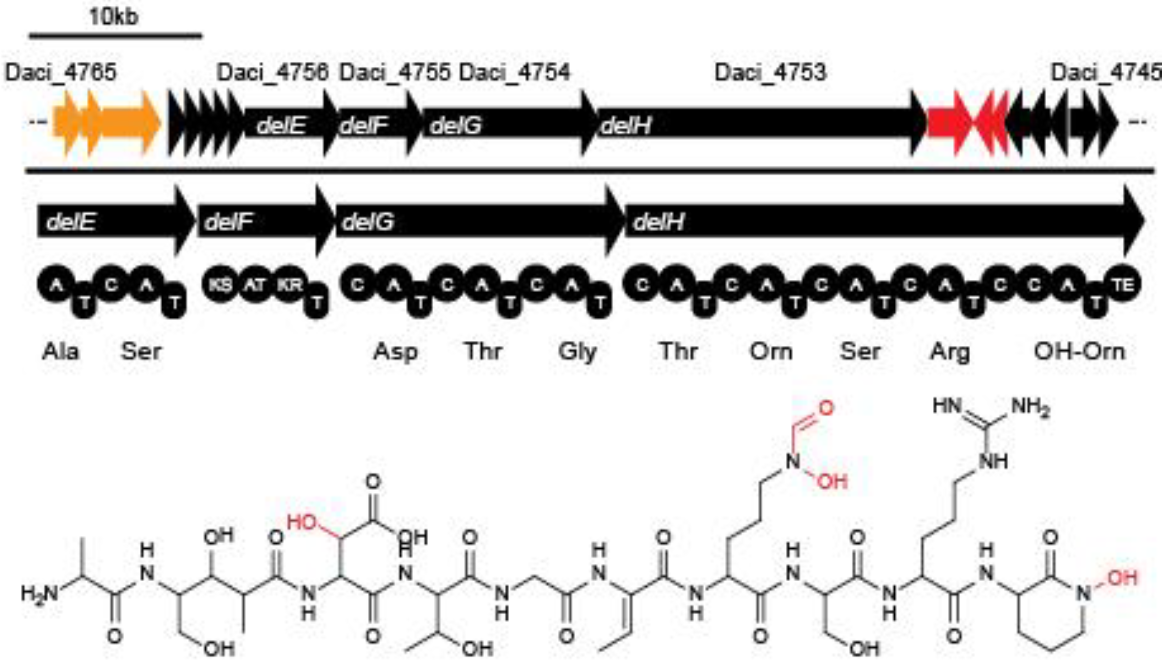
http://2013.igem.org/Team:Heidelberg/Project/Delftibactin
Proteobacteria
Betaproteobacteria
Burkholderiales
Comamonadaceae
Delftia
Our Workflow:

Each step will be outlined before you reach it with a description of the apps, parameters and results.
We'll start with a number of raw reads, clean them up and do a little taxonomy to figure out what we might be able to assemble. Next, we'll assemble the reads using 4 different methods and pick out the best assembly. Then we'll take those reads and sort them by genome and assess the quality of those genomes. We'll select the high quality genomes to annotate and insert into phylogenetic trees to find relatives.
After completing this narrative, students will be able to:
Sample 1: Metagenome generated from fracture fluid collected July 12-27, 2012 from a borehole located on the 26 level of Beatrix Gold Mine (Welkom, South Africa) 1,339 m below land surface.
Bio Sample: SAMN04419121; Sample name: Be326_2012_DNA_MF; SRA: SRS1792548 Link to more information: https://www.ncbi.nlm.nih.gov/biosample/SAMN04419121
Why did I pick this sample?
Delftia acidovorans and Cupriavidus metallidurans make up 90% of the bacteria found in biofilms on gold nuggets (1). This sample is from a gold mine, so it would be interesting to see if Delftia is also present here. Additionally, the Krona plot indicated that there were some (unassembled) sequences that resembled Delftia.
Sample 2: Hydraulically fractured gas well metagenomes fluid from sample at timepoint_82
Bio Sample: SAMN04417545; Sample name: Timepoint_82; SRA: SRS1256393 Link to more information: https://www.ncbi.nlm.nih.gov/biosample/SAMN04417545
Why did I pick this sample?
Delftia is often found in soil and water. It is capable of using a diverse array of carbon sources as well. (That's why so many studies focus on it's use for bioremediation.) In this case the Krona plot indicated a small portion of potential Delftia sequences.
Sample 3: Subsurface sediment microbial communities from gas well in Oklahoma, United States - OK STACK MC-FT3-sol metagenome
Bio Sample: SAMN09199659; SRA: SRS3667068; DOE Joint Genome Institute: Gp0290895 Link to more information: https://www.ncbi.nlm.nih.gov/biosample/SAMN09199659
Why did I pick this sample?
I stumbled upon this sample while I was searching through the database. The Krona plot showed a much greater proportion of genes that could potentially be from Delftia, and I couldn't skip it. Delftia is often found in soil and/or sediment.
Sample 4: Metagenome of iron plaque on rice root from As contaminated paddy soil, sample from Yanhong
Bio Sample: SAMN07211852; Sample name: YanhongMeta01; SRA: SRS2392319 Link to more information: https://www.ncbi.nlm.nih.gov/biosample/SAMN07211852
Why did I pick this sample?
I choose this sample because it brings together soil and aquatic environments that Delftia can be found in and is associated with heavy metals. The Krona plot indicated some Delftia-associated sequences are present from several species of Delftia.
Sample 5: Peat soil microbial communities from Stordalen Mire, Sweden - IR.F.S.T-25
Bio Sample: SAMN09201211; SRA: SRS3568559,DOE Joint Genome Institute: Gp0256443, Link to more information: https://www.ncbi.nlm.nih.gov/sra/SRX4415252[accn]
Why did I pick this sample?
I picked this sample because soil is one of the common sources of Delftia. The Krona plot looked promising as well for both Delftia acidovorans and Delftia tsuruhatensis.
The data I'm using comes from publicly available datasets available from NCBI's Sequence Read Archive (SRA).
The sample you need to upload should be a metagenomic sample listed as a WGS having paired reads. Your sample cannot be more than 20G bases or you'll need to import it through Globus (see this link for more information). You can also check out this really informative narrative for an in depth view of how to upload data from other sources: https://narrative.kbase.us/narrative/48493
App: Import SRA File as Reads from Web
Timing: 1-5 hours depending on the size of the file and queue time. (Its often helpful to run this in the morning so it uploads and then you can set up the assembly to run overnight or over the weekend.)
View Configure:
SRA URL: Use the first link from the "Reads Access" page and paste it into this block.
Reads Object Name: this will be what your sample is called once it's been uploaded into KBase. Make sure its something you'll be able to remember and follow the workflow with.
Sequencing Technology: Select the sequencing technology that was used to call the reads.
Single Genome: These reads are all metagenomes, so you don't need to select this box.
Results: Your reads should appear in the Data panel to the left. Before proceding, double check that they are a PairedEndLibrary. If they are a SingleEndLibrary, you'll need to select a different sample, because your assembly won't work with a SingleEndLibrary. In the results panel itself you'll see some stats from the reads including the number of reads, quality score mean and mean read length.

| Created Object Name | Type | Description |
|---|---|---|
| Subsurface_gold_mine_reads | PairedEndLibrary | Imported Reads |
| Created Object Name | Type | Description |
|---|---|---|
| Hydraulic_fracture_well_fluid_raw_reads | PairedEndLibrary | Imported Reads |
| Created Object Name | Type | Description |
|---|---|---|
| Subsurface_gas_well_reads | PairedEndLibrary | Imported Reads |
| Created Object Name | Type | Description |
|---|---|---|
| Rice_root_iron_plaque_reads | PairedEndLibrary | Imported Reads |
| Created Object Name | Type | Description |
|---|---|---|
| Peat_soil_raw_reads | PairedEndLibrary | Imported Reads |
You've uploaded your data, great! Now you have to check the quality of the reads. First assess quality with FastQC. Then, if you need to, trim the reads and assess quality again to make sure the low quality reads have been removed from the sample.
Apps:
Timing: 5-20 minutes depending on queue and the number of reads
Assess Read Quality with FastQC View Configure:
Results: This app will give you a full report of the quality of your reads. The report will have two pages, for the forward and reverse reads in the PairedEndLibrary. Here we'll be focusing on the Per Base Sequence Quality, but the rest of the report offers a bunch of useful information about our library.
For a more in depth look at understanding your FastQC report, check out the manual here: https://dnacore.missouri.edu/PDF/FastQC_Manual.pdf
Trim Reads with Trimmommatic View Configure
Read library or set: Select the library you want to trim.
Parameters: This section covers different trimming parameters specific to removing adapters, croping the sequences, and the quality thresholds required to trim a read. In this workflow, I'm going to leave them all as defaults, but you can learn more about them in the App Info page and in the KBase App catalog.
Output library name: Make sure to specify that this library has been trimmed so you can tell the two libraries apart later.
Once you've trimmed the read library, reassess the quality of the PairedEndLibrary using FastQC.
Results: A FastQC Report that details the quality of the reads you've submitted and possibly trimmed libraries.

Q1 Looking at the results of the FastQC app, which reads set(s) would you choose to trim and re-assess the quality of? Why did you pick that/those set(s)?

| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_reads_trimmed_paired | PairedEndLibrary | Trimmed Reads |
| subsurface_gold_mine_reads_trimmed_unpaired_fwd | SingleEndLibrary | Trimmed Unpaired Forward Reads |
| subsurface_gold_mine_reads_trimmed_unpaired_rev | SingleEndLibrary | Trimmed Unpaired Reverse Reads |
| Created Object Name | Type | Description |
|---|---|---|
| Peat_soil_reads_trimmed_paired | PairedEndLibrary | Trimmed Reads |
| Peat_soil_reads_trimmed_unpaired_fwd | SingleEndLibrary | Trimmed Unpaired Forward Reads |
| Peat_soil_reads_trimmed_unpaired_rev | SingleEndLibrary | Trimmed Unpaired Reverse Reads |
Before we assemble these libraries, it will be helpful to get an idea of what's present in our samples and at what abundance. KBase has two apps to do this, Kaiju and GOTTCHA2.
Apps:
Timing: 20 mins-2 hours depending on queue and number of reads you're running
1. Classify Taxonomy of Metagenomic Reads with Kaiju
This app translates reads into proteins and uses those sequences to identify what's present or possibly present in the sample.
View Configure:
Results: Your results will be a series of tables showing the breakdown of your sample beginning with the phyla and ending at species. The tail includes everything that is present below the low abundance filter.
2. Classify Taxonomy of Metagenomic Reads with GOTTCHA2
Unlike Kaiju, this app shows relative abundance based on unique nucleotide sequences from RefSeq.
View Configure:
Results: There are three ways you can view the results from GOTTCHA2. The first is as a table showing the classification of your reads, some statistics regarding their abundance and their relative abundance. The second is as a phylogenetic tree showing the relationships of the different taxa identified in your sample. The third layout is as a Krona plot, an interactive plot that displays relative abundance and phylogenetic relationships. Clicking on a phylum will zoom in to show the classes within it. How far you can zoom down depends on the sample and the unique sequences in it.
These first two runs of Kaiju include the first 4 libraries run as one first with NCBI BLAST as the database, then with RefSeq as the database. The following 4 examples show the libraries individually with different settings to increase the proportion of the library represented.

| subsurface_gold_mine_reads_trimmed_paired |
| Rice_root_iron_plaque_reads |
| Subsurface_gas_well_reads |
| Hydraulic_fracture_well_fluid_raw_reads |
| ALL |
| subsurface_gold_mine_reads_trimmed_paired |
| Rice_root_iron_plaque_reads |
| Subsurface_gas_well_reads |
| Hydraulic_fracture_well_fluid_raw_reads |
| ALL |
These runs of Kaiju are broken down by sample and use the NCBI BLAST database. They are run on two subsamples.
| subsurface_gold_mine_reads_trimmed_paired |
| ALL |
Q2 Do the two subsamples vary much? Is this expected or unexpected and why?
| Hydraulic_fracture_well_fluid_raw_reads |
| ALL |
| Subsurface_gas_well_reads |
| ALL |
| Rice_root_iron_plaque_reads |
| ALL |
| Peat_soil_reads_trimmed_paired |
| ALL |
This first run of GOTTCHA2 includes ALL read libraries as one sample. The 4 runs afterwards show the samples individually.
| subsurface_gold_mine_reads_trimmed_paired |
| Rice_root_iron_plaque_reads |
| Subsurface_gas_well_reads |
| Hydraulic_fracture_well_fluid_raw_reads |
| subsurface_gold_mine_reads_trimmed_paired |
Q3: Open the Krona plot from this sample. Viruses make up what percent of the sample?
| Hydraulic_fracture_well_fluid_raw_reads |
| Subsurface_gas_well_reads |
Q4: What is the relative abundance of Betaproteobacteria in this sample?
| Rice_root_iron_plaque_reads |
| Peat_soil_reads_trimmed_paired |
Q5: Which sample(s) look the most promising based on the taxonomy results from GOTTCHA2?
Alright, you've made it this far. This step takes the longest, so you may want to set it up to run overnight or over the weekend. This step will take our read libraries and line them up into longer sequences called contigs. Later we'll sort these contigs based on what genomes they came from. We'll be using three different apps to generate 4 different sets of contigs.
Apps:
Timing: hours to days (One of my assemblies below ran for almsot 4 days.)
1. Assemble Reads with MetaSPAdes
View Configure:
2. Assemble Reads with MEGAHIT (run 2x)
View Configure:
3. Assemble Reads with IDBA-UD
View Configure:
Results: Regardless of the assembly you use, you'll get the same result; a QUAST report. This report details the major features of your assembly and important statistics, such as the length of the longest read, the number of reads longer than 1,000,000 bases, the N50 and L50. In the next step we'll compare these statistics and receive a visual representation of the key parts of this report. For now, take note of which assembly contains the most base pairs.

| Created Object Name | Type | Description |
|---|---|---|
| Subsurface_gold_mine_IDBA.contigs | Assembly | Assembled contigs |

| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_meta_sensitive_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_meta_large_MEGAHIT.assembly | Assembly | Assembled contigs |

| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| hydraulic_fracture_well_IDBA.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| meta_sensitive_hydraulic_well_2_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| meta_large_hydraulic_fracture_well_2_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| hydraulic_fracture_well_SPAdes.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_IDBA.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_meta_sensitive_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_reads_meta_large_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_SPAdes.contigs | Assembly | Assembled contigs |
Q6 How long did it take (including the queue time) to assemble this reads dataset with metaSPAdes?
| Created Object Name | Type | Description |
|---|---|---|
| rice_root_iron_plaque_IDBA.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| rice_root_iron_plaque_meta_sensitive_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| rice_root_iron_plaque_meta_large_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| Peat_soil_IDBA.contigs | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| peat_soil_meta_sensitive_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| peat_soil_meta_large_MEGAHIT.assembly | Assembly | Assembled contigs |
| Created Object Name | Type | Description |
|---|---|---|
| peat_soil_SPAdes.contigs | Assembly | Assembled contigs |
Success! You've created 4 different assemblies from your metagenomic data. Now, you need to pick the best to sort out all those contigs into genomes. That step, sorting the contigs into different genome bins is called binning. Each bin will represent a single genome, but more on that later.
App: Compare Assembled Contig Distributions
Timing: 5-10 mins.
View Configure:
All you need to add here are your different assemblies from above. For example, in the first run I'll compare the Subsurface_gold_mine_meta_sensitive_MEGAHIT.assembly, subsurface_gold_mine_meta_large_MEGAHIT.assembly, subsurface_gold_mine_IDBA.contigs, and subsurface_gold_mine_SPAdes to determine which is the best to use moving forward.
The Results: A report showing the different statistics from each assembly. Some key features of this report include:
A good assembly will have:
A bad assembly will have:

| subsurface_gold_mine_meta_sensitive_MEGAHIT.assembly |
| subsurface_gold_mine_SPAdes.contigs |
| Subsurface_gold_mine_IDBA.contigs |
| subsurface_gold_mine_meta_large_MEGAHIT.assembly |
Q7a Which assembly would you use for binning contigs from this sample? Why did you pick that assembly?
Q7b Which is the worst choice for binning contigs? Why is it the worst?
| meta_large_hydraulic_fracture_well_2_MEGAHIT.assembly |
| meta_sensitive_hydraulic_well_2_MEGAHIT.assembly |
| hydraulic_fracture_well_SPAdes.contigs |
| hydraulic_fracture_well_IDBA.contigs |
Q8 Which assembly should I use to bin the contigs from the hydraulic fracture well fluid sample? Why?
| subsurface_gas_well_SPAdes.contigs |
| subsurface_gas_well_reads_meta_large_MEGAHIT.assembly |
| rice_root_iron_plaque_meta_sensitive_MEGAHIT.assembly |
| subsurface_gas_well_IDBA.contigs |
Q9a Which assembly is the best for binning the contigs from the subsurface gas well sample? Is there more than one option? If so, which one would you use and why?
Q9b Which assembly can you rule out?
| rice_root_iron_plaque_IDBA.contigs |
| rice_root_iron_plaque_SPAdes.contigs |
| rice_root_iron_plaque_meta_sensitive_MEGAHIT.assembly |
| rice_root_iron_plaque_meta_large_MEGAHIT.assembly |
Q10 Which assembly should I use to bin the contigs from the rice root iron plaque sample? Why?
| peat_soil_SPAdes.contigs |
| Peat_soil_IDBA.contigs |
| peat_soil_meta_large_MEGAHIT.assembly |
| peat_soil_meta_sensitive_MEGAHIT.assembly |
Q11 Which assembly should I use to bin the contigs from the peat soil sample? Why?
Alright, you've picked the best assembly, now you'll sort all the contigs into bins that each represent a single genome. This step is called binning the contigs.
App: Bin Contigs using MaxBin2
Timing: 2+ hours depending on the number of contigs and bins present in your sample.
View Configure:
Assembly Object: Put your best assembly here.
Read Library: This is the library the contigs were generated from. If you needed to trim your read library, use the trimmed reads.
Probability Threshold: The confidence the alrogrithm must have for a contig to be placed within a bin. If a contig falls below this cutoff, then it will be left as unclassified. The default is 0.8.
Marker Set: MaxBin2 can bin both bacterial and archaeal genomes. In this case we're only looking at bacteria, so keep it set to the bacterial marker gene set.
Minimum contig length: Any contigs shorter than this will be ignored when binning. 1000 is the default, but above we set our contig minimum length at 2000 so we can increase this to 2000 or leave it as is, since we shouldn't have any contigs shorter than 2000 bases.
Results: The output from this app opens in a new section. The first panel lists the number of bins (and maximum number of genomes) and nucleotides included in all the contigs. The second tab offers some detail about the different bins including marker completenes, GC content, the number of contigs in each bin and their total length. To see information about the individual contigs in a bin, click the bulleted list icon for that bin or the graph beside it. However, these results tell you nothing about the quality of the bins, they could be highly contaminated or contain multiple copies of the same set of genes.

| subsurface_gold_mine_reads_trimmed_paired |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_meta_sensitive_bins | BinnedContigs | BinnedContigs from MaxBin2 |
Q12 Delftia acidovorans has a GC content of 66.7%. Based on GC content alone, do any of these bins match D. acidovorans?
| Hydraulic_fracture_well_fluid_raw_reads |
| Created Object Name | Type | Description |
|---|---|---|
| hydraulic_fracture_well_fluid_meta_large_bins | BinnedContigs | BinnedContigs from MaxBin2 |
Q13 What is unusual about the bins from the hydraulic fracture well fluid sample?
Hint: look at the bins tab.
| Subsurface_gas_well_reads |
| Created Object Name | Type | Description |
|---|---|---|
| Subsurface_gas_well_spade_bins | BinnedContigs | BinnedContigs from MaxBin2 |
| Rice_root_iron_plaque_reads |
| Created Object Name | Type | Description |
|---|---|---|
| Rice_root_iron_plaque_SPAdes_bins | BinnedContigs | BinnedContigs from MaxBin2 |
| Peat_soil_reads_trimmed_paired |
| Created Object Name | Type | Description |
|---|---|---|
| Peat_soil_meta_sens_contigs | BinnedContigs | BinnedContigs from MaxBin2 |
You should now have a bunch of different bins. Each bin represents a single genome (in theory). In this step we'll check these bins for their completeness, contamination and any duplicates using CheckM.
App: Assess Genome Quality with CheckM
Timing: Depends on the number of bins in your sample and the reference tree you pick
View Configure
Input Assembly, Genome or BinnedContigs: Add in your set of bins from the last step here.
Reference Tree: You can either select the full tree or reduced tree to compare your bins to. The full tree takes longer, but is recommended for a better understanding of what each bin represents. However, if you're tight on time the reduced tree is fine since we'll be generating a species tree to determine close relatives of our assemblies later.
Save all Plots: Save will allow you to download a .zip file of the resulting genome quality plots. Don't save will not.
Results: CheckM will give you two forms of the same report, a graphic version and a table. I think the table is easier to understand, so that's what I'll be covering here. The first column shows the bin name. They're all just numbered bins at this point, but you can rename them later if you want. The second column shows the lineage of the markers present in that bin. Some will be more specific than others, depending on the bin, its completeness and contamination. Number of genomes is the number of genomes used to create the marker set, and number of markers is the number of markers generated. These markers are unique and are expected to occur only once in the genome, replicates indicate contamination. The columns 0 through 5+ indicate the additional copies of these marker genes and are used to calculate contamination. Be aware that contamination is an underestimate in this app.
The last two columns indicate the completeness and contamination of your genome as percents. High quality genomes are over 90% complete with less than 5% contamination. However, since we're just looking to ID if Delftia is present, I'm using any genomes over 75% complete with less than 5% contamination. If an assembly falls outside this range, but looks promising, you can keep it, but be sure to note that it's a low quality assembly.
Write out a list of all the bins you want to keep, it will be useful in the next step.

Q14a Which bins would you select to keep working with?
Q14b Why did you pick those bins?
Q15 Are any of these bins high enough quality to keep working with? If so, which one(s)?

| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_spade_CheckM_HQ_bins.BinnedContigs | BinnedContigs | HQ BinnedContigs subsurface_gas_well_spade_CheckM_HQ_bins.BinnedContigs |
Q16 This one looks more promising! Which bins would you choose to extract and are there any that look like Delftia?
Up above you checked the quality of all your bins and picked out all the ones that were over 75% complete and less than 5% contaminated. Now you're going to separate them from the contaminated and incomplete assemblies so you just have to work with them.
App: Extract Bins as Assemblies from BinnedContigs
Timing: Depends on the number of bins you're extracting, in general ~10 minutes or so
View Configure:
Binned Contigs: Select the binned contigs set you put into CheckM above. Once you add it the data will automatically fill into the lower Parameters section.
Bin Names Available for Extraction: There is a green plus on the right side of all your bins. Click it to select the ones you want to save as assemblies. They will appear in the lower table. Once you're done, double check that they're all there and that you got the right ones.
Assembly Name Suffix: Your bins will be renamed with this added. It should be a descriptive suffix so you can tell them apart, because any extracted bins will start with Bin###.fasta
AssemblySet Name: This will be the name of your assembly set that contains all your extracted bins. Again, it should be named something descriptive. For example, I named the first assembly set : Subsurface_gold_mine_extracted_bins.AssemblySet
If you have just one bin to extract your results will just be an assembly, because an AssemblySet needs to include 2 or more assemblies. This won't produce an error message.
Results: Your results will include a table of the different assemblies and a note that the job finished successfully.

| Bin.009.fasta |
| Bin.011.fasta |
| Bin.013.fasta |
| Bin.015.fasta |
| Bin.017.fasta |
| Bin.019.fasta |
| Bin.020.fasta |
| Bin.022.fasta |
| Bin.033.fasta |
| Bin.034.fasta |
| Bin.035.fasta |
| Bin.036.fasta |
| Bin.040.fasta |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gold_mine_extracted_bins.AssemblySet | AssemblySet | Assembly set of extracted assemblies |
| Bin.009.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.011.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.013.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.015.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.017.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.019.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.020.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.022.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.033.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.034.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.035.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.036.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.040.fasta_subsurface_gold_mine_assembly | Assembly | Assembly object of extracted contigs |
| Bin.004.fasta |
| Created Object Name | Type | Description |
|---|---|---|
| Bin.004.fasta_assembly | Assembly | Assembly object of extracted contigs |
| Bin.003.fasta |
| Bin.005.fasta |
| Bin.013.fasta |
| Created Object Name | Type | Description |
|---|---|---|
| subsurface_gas_well_spade_extracted_bins.AssemblySet | AssemblySet | Assembly set of extracted assemblies |
| Bin.003.fasta_assembly | Assembly | Assembly object of extracted contigs |
| Bin.005.fasta_assembly | Assembly | Assembly object of extracted contigs |
| Bin.013.fasta_assembly | Assembly | Assembly object of extracted contigs |
| Bin.001.fasta |
| Bin.002.fasta |
| Bin.005.fasta |
| Bin.008.fasta |
| Bin.009.fasta |
| Bin.010.fasta |
| Bin.012.fasta |
| Bin.014.fasta |
| Bin.015.fasta |
| Bin.016.fasta |
| Bin.026.fasta |
| Created Object Name | Type | Description |
|---|---|---|
| rice_root_iron_plaque_extracted_bins.AssemblySet | AssemblySet | Assembly set of extracted assemblies |
| Bin.001.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.002.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.005.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.008.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.009.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.010.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.012.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.014.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.015.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.016.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.026.fasta_rice_root_plaque_assembly | Assembly | Assembly object of extracted contigs |
| Bin.001.fasta |
| Bin.007.fasta |
| Bin.009.fasta |
| Created Object Name | Type | Description |
|---|---|---|
| peat_soil_extracted_bins.AssemblySet | AssemblySet | Assembly set of extracted assemblies |
| Bin.001.fastapeat_soil_assembly | Assembly | Assembly object of extracted contigs |
| Bin.007.fastapeat_soil_assembly | Assembly | Assembly object of extracted contigs |
| Bin.009.fastapeat_soil_assembly | Assembly | Assembly object of extracted contigs |
You now have a set of mostly-whole, mostly-uncontaminated genomes from your sample. Now you'll use RAST to identify genes in these assemblies.
App: Annotate Multiple Microbial Assemblies (RAST)
Timing: Depends on the number of assemblies and their size. I'd estimate it takes about 10 mins an assembly.
View Configure:
Assemblies/AssemblySets: Here is where you add the AssemblySet you generated in the last step. You could add your assemblies individually, but it's easier to add them all as one set.
Domain and Genetic Code: Both should be set for bacteria, since D. acidovorans is a bacteria.
Call Buttons: By default most of these are checked. For something like a genome assembly, it's good to grab more features than we need in case we need them for a future study.
Results: Your results from RAST are fairly simple. You'll get objects for each assembly and one set of all assemblies annotated together. The summary will give you a short description of what was annotated in each genome and if the annotation was a success. Check through the summary to make sure none of the assemblies failed. Sometimes one will fail, but the app will still give you a success message. If it does fail, try annotating that assembly alone using the app: Annotate Microbial Assembly with different settings.

| subsurface_gold_mine_extracted_bins.AssemblySet |
The RAST algorithm was applied to annotating a genome sequence comprised of 252 contigs containing 3023957 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3829 new features were called, of which 305 are non-coding. Output genome has the following feature types: Coding gene 3524 Non-coding crispr_array 3 Non-coding crispr_repeat 22 Non-coding crispr_spacer 19 Non-coding repeat 214 Non-coding rna 47 Overall, the genes have 1296 distinct functions. The genes include 1474 genes with a SEED annotation ontology across 814 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.036.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 40 contigs containing 1396467 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 2005 new features were called, of which 202 are non-coding. Output genome has the following feature types: Coding gene 1803 Non-coding crispr_array 3 Non-coding crispr_repeat 42 Non-coding crispr_spacer 41 Non-coding repeat 65 Non-coding rna 51 Overall, the genes have 749 distinct functions. The genes include 798 genes with a SEED annotation ontology across 558 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.011.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 41 contigs containing 1401304 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 1470 new features were called, of which 91 are non-coding. Output genome has the following feature types: Coding gene 1379 Non-coding repeat 44 Non-coding rna 47 Overall, the genes have 432 distinct functions. The genes include 594 genes with a SEED annotation ontology across 313 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.013.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 45 contigs containing 3965585 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3523 new features were called, of which 125 are non-coding. Output genome has the following feature types: Coding gene 3398 Non-coding crispr_array 1 Non-coding crispr_repeat 6 Non-coding crispr_spacer 5 Non-coding repeat 68 Non-coding rna 45 Overall, the genes have 1237 distinct functions. The genes include 1910 genes with a SEED annotation ontology across 774 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.022.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 241 contigs containing 3603719 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3603 new features were called, of which 413 are non-coding. Output genome has the following feature types: Coding gene 3190 Non-coding crispr_array 1 Non-coding crispr_repeat 13 Non-coding crispr_spacer 12 Non-coding repeat 351 Non-coding rna 36 Overall, the genes have 1073 distinct functions. The genes include 1723 genes with a SEED annotation ontology across 675 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.033.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 90 contigs containing 1414679 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3067 new features were called, of which 87 are non-coding. Output genome has the following feature types: Coding gene 2980 Non-coding repeat 51 Non-coding rna 36 Overall, the genes have 140 distinct functions. The genes include 277 genes with a SEED annotation ontology across 103 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.034.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 84 contigs containing 3078190 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3001 new features were called, of which 120 are non-coding. Output genome has the following feature types: Coding gene 2881 Non-coding repeat 75 Non-coding rna 45 Overall, the genes have 1088 distinct functions. The genes include 1490 genes with a SEED annotation ontology across 733 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.035.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 102 contigs containing 4104362 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4141 new features were called, of which 290 are non-coding. Output genome has the following feature types: Coding gene 3851 Non-coding crispr_array 1 Non-coding crispr_repeat 49 Non-coding crispr_spacer 48 Non-coding repeat 135 Non-coding rna 57 Overall, the genes have 1484 distinct functions. The genes include 1918 genes with a SEED annotation ontology across 875 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.009.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 6 contigs containing 574605 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 687 new features were called, of which 55 are non-coding. Output genome has the following feature types: Coding gene 632 Non-coding repeat 10 Non-coding rna 45 Overall, the genes have 246 distinct functions. The genes include 337 genes with a SEED annotation ontology across 204 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.017.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 22 contigs containing 1075402 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 1247 new features were called, of which 56 are non-coding. Output genome has the following feature types: Coding gene 1191 Non-coding repeat 7 Non-coding rna 49 Overall, the genes have 361 distinct functions. The genes include 520 genes with a SEED annotation ontology across 278 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.019.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 87 contigs containing 2205918 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 2368 new features were called, of which 131 are non-coding. Output genome has the following feature types: Coding gene 2237 Non-coding crispr_array 1 Non-coding crispr_repeat 5 Non-coding crispr_spacer 4 Non-coding repeat 87 Non-coding rna 34 Overall, the genes have 1438 distinct functions. The genes include 1224 genes with a SEED annotation ontology across 851 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.020.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 339 contigs containing 3344248 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3842 new features were called, of which 171 are non-coding. Output genome has the following feature types: Coding gene 3671 Non-coding crispr_array 2 Non-coding crispr_repeat 41 Non-coding crispr_spacer 39 Non-coding repeat 48 Non-coding rna 41 Overall, the genes have 1737 distinct functions. The genes include 1757 genes with a SEED annotation ontology across 1014 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.040.fasta_subsurface_gold_mine_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 85 contigs containing 2402219 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 2888 new features were called, of which 310 are non-coding. Output genome has the following feature types: Coding gene 2578 Non-coding crispr_array 2 Non-coding crispr_repeat 65 Non-coding crispr_spacer 63 Non-coding repeat 130 Non-coding rna 50 Overall, the genes have 1212 distinct functions. The genes include 1265 genes with a SEED annotation ontology across 788 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.015.fasta_subsurface_gold_mine_assembly succeeded!
Q17 Did all assembled genomes annotate correctly?
| Bin.004.fasta_assembly |
| Created Object Name | Type | Description |
|---|---|---|
| Bin.004.fasta_assembly.RAST | Genome | Annotated genome |
| hydraulic_fracture_well_Bin.004_annotated_assembly | GenomeSet | Genome Set |
The RAST algorithm was applied to annotating a genome sequence comprised of 75 contigs containing 3624967 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4069 new features were called, of which 680 are non-coding. Output genome has the following feature types: Coding gene 3389 Non-coding crispr_array 3 Non-coding crispr_repeat 247 Non-coding crispr_spacer 244 Non-coding repeat 129 Non-coding rna 57 Overall, the genes have 2271 distinct functions. The genes include 1778 genes with a SEED annotation ontology across 1273 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.004.fasta_assembly succeeded!
| subsurface_gas_well_spade_extracted_bins.AssemblySet |
| Created Object Name | Type | Description |
|---|---|---|
| Bin.003.fasta_assembly.RAST | Genome | Annotated genome |
| Bin.013.fasta_assembly.RAST | Genome | Annotated genome |
| Bin.005.fasta_assembly.RAST | Genome | Annotated genome |
| subsurface_gas_well_extracted_bins_annotated | GenomeSet | Genome Set |
The RAST algorithm was applied to annotating a genome sequence comprised of 85 contigs containing 4601080 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4522 new features were called, of which 158 are non-coding. Output genome has the following feature types: Coding gene 4364 Non-coding repeat 104 Non-coding rna 54 Overall, the genes have 2996 distinct functions. The genes include 1606 genes with a SEED annotation ontology across 1174 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.003.fasta_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 191 contigs containing 4467743 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4441 new features were called, of which 90 are non-coding. Output genome has the following feature types: Coding gene 4351 Non-coding repeat 49 Non-coding rna 41 Overall, the genes have 2662 distinct functions. The genes include 1851 genes with a SEED annotation ontology across 1186 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.013.fasta_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 127 contigs containing 4858083 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4781 new features were called, of which 120 are non-coding. Output genome has the following feature types: Coding gene 4661 Non-coding repeat 46 Non-coding rna 74 Overall, the genes have 2211 distinct functions. The genes include 1980 genes with a SEED annotation ontology across 1116 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.005.fasta_assembly succeeded!
| rice_root_iron_plaque_extracted_bins.AssemblySet |
| Created Object Name | Type | Description |
|---|---|---|
| Bin.012.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.010.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.009.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.008.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.015.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.014.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.001.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.026.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.005.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.002.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| Bin.016.fasta_rice_root_plaque_assembly.RAST | Genome | Annotated genome |
| rice_root_iron_plaque_annotated_extracted_bins | GenomeSet | Genome Set |
The RAST algorithm was applied to annotating a genome sequence comprised of 328 contigs containing 4422408 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4368 new features were called, of which 84 are non-coding. Output genome has the following feature types: Coding gene 4284 Non-coding repeat 20 Non-coding rna 64 Overall, the genes have 2995 distinct functions. The genes include 1967 genes with a SEED annotation ontology across 1474 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.012.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 21 contigs containing 3884316 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3412 new features were called, of which 50 are non-coding. Output genome has the following feature types: Coding gene 3362 Non-coding repeat 10 Non-coding rna 40 Overall, the genes have 1625 distinct functions. The genes include 1522 genes with a SEED annotation ontology across 854 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.010.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 34 contigs containing 3177223 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3105 new features were called, of which 113 are non-coding. Output genome has the following feature types: Coding gene 2992 Non-coding repeat 56 Non-coding rna 57 Overall, the genes have 1576 distinct functions. The genes include 1395 genes with a SEED annotation ontology across 790 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.009.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 64 contigs containing 6397280 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 6032 new features were called, of which 81 are non-coding. Output genome has the following feature types: Coding gene 5951 Non-coding repeat 31 Non-coding rna 50 Overall, the genes have 3129 distinct functions. The genes include 2588 genes with a SEED annotation ontology across 1458 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.008.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 123 contigs containing 5324791 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 5069 new features were called, of which 70 are non-coding. Output genome has the following feature types: Coding gene 4999 Non-coding repeat 29 Non-coding rna 41 Overall, the genes have 2933 distinct functions. The genes include 2304 genes with a SEED annotation ontology across 1385 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.015.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 62 contigs containing 6676535 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 6156 new features were called, of which 117 are non-coding. Output genome has the following feature types: Coding gene 6039 Non-coding repeat 51 Non-coding rna 66 Overall, the genes have 2270 distinct functions. The genes include 2399 genes with a SEED annotation ontology across 996 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.014.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 27 contigs containing 3919630 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3807 new features were called, of which 70 are non-coding. Output genome has the following feature types: Coding gene 3737 Non-coding repeat 19 Non-coding rna 51 Overall, the genes have 2059 distinct functions. The genes include 1896 genes with a SEED annotation ontology across 1163 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.001.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 560 contigs containing 3298417 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3683 new features were called, of which 27 are non-coding. Output genome has the following feature types: Coding gene 3656 Non-coding repeat 2 Non-coding rna 25 Overall, the genes have 2252 distinct functions. The genes include 1440 genes with a SEED annotation ontology across 955 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.026.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 19 contigs containing 3296343 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3242 new features were called, of which 52 are non-coding. Output genome has the following feature types: Coding gene 3190 Non-coding repeat 8 Non-coding rna 44 Overall, the genes have 2006 distinct functions. The genes include 1677 genes with a SEED annotation ontology across 1085 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.005.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 216 contigs containing 4630571 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4695 new features were called, of which 71 are non-coding. Output genome has the following feature types: Coding gene 4624 Non-coding repeat 19 Non-coding rna 52 Overall, the genes have 3392 distinct functions. The genes include 2041 genes with a SEED annotation ontology across 1681 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.002.fasta_rice_root_plaque_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 131 contigs containing 4160543 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 4100 new features were called, of which 79 are non-coding. Output genome has the following feature types: Coding gene 4021 Non-coding repeat 35 Non-coding rna 44 Overall, the genes have 2272 distinct functions. The genes include 2144 genes with a SEED annotation ontology across 1142 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.016.fasta_rice_root_plaque_assembly succeeded!
| peat_soil_extracted_bins.AssemblySet |
| Created Object Name | Type | Description |
|---|---|---|
| Bin.001.fastapeat_soil_assembly.RAST | Genome | Annotated genome |
| Bin.007.fastapeat_soil_assembly.RAST | Genome | Annotated genome |
| Bin.009.fastapeat_soil_assembly.RAST | Genome | Annotated genome |
| Peat_soil_extracted_bins_annotated | GenomeSet | Genome Set |
The RAST algorithm was applied to annotating a genome sequence comprised of 173 contigs containing 1950668 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 2844 new features were called, of which 218 are non-coding. Output genome has the following feature types: Coding gene 2626 Non-coding repeat 180 Non-coding rna 38 Overall, the genes have 774 distinct functions. The genes include 905 genes with a SEED annotation ontology across 468 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.001.fastapeat_soil_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 186 contigs containing 2021755 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 2229 new features were called, of which 70 are non-coding. Output genome has the following feature types: Coding gene 2159 Non-coding repeat 26 Non-coding rna 44 Overall, the genes have 877 distinct functions. The genes include 1117 genes with a SEED annotation ontology across 626 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.007.fastapeat_soil_assembly succeeded! The RAST algorithm was applied to annotating a genome sequence comprised of 492 contigs containing 2734491 nucleotides. No initial gene calls were provided. Standard features were called using: glimmer3; prodigal. A scan was conducted for the following additional feature types: rRNA; tRNA; selenoproteins; pyrrolysoproteins; repeat regions; crispr. The genome features were functionally annotated using the following algorithm(s): Kmers V2; Kmers V1; protein similarity. In addition to the remaining original 0 coding features and 0 non-coding features, 3214 new features were called, of which 138 are non-coding. Output genome has the following feature types: Coding gene 3076 Non-coding repeat 108 Non-coding rna 30 Overall, the genes have 1139 distinct functions. The genes include 1365 genes with a SEED annotation ontology across 736 distinct SEED functions. The number of distinct functions can exceed the number of genes because some genes have multiple functions. Bin.009.fastapeat_soil_assembly succeeded!
Now that you've annotated your assemblies, it's time to figure out what they are!
App: GTDB-Tk classify
Timing: about 30 minutes, depending on the number of assemblies and the queue time
View Configure:
Assembly Input: Add in your annotated assembly set from above.
Minimum Alignment Percent: This will filter out genomes with an insufficient percentage of AAs in the MSA generated by the app. The default is 10, if you want to increase the specificity all you need to do is increase this percentage. In my runs below I've kept the default as it is.
Results: The results of this app are across 4 tabs. The first tab is a table for bacteria and the second shows the same table for archaea. The first column indicates the bin and the second indicates the classification of that bin based on GenBank and RefSeq databases. The middle columns offer information about how this classification was determined. The right-most column is also important, since it will note any concerns about your genome. For example, if it contains high levels of contamination.
Q18 Were any of the assembled genomes from Delftia? If not, what did you find instead?
Q19 Was the assembled genome from Delftia? If not, what was it instead?
Lastly, I'll identify close relatives to my bins just to establish some addtional phylogenetic context for them.
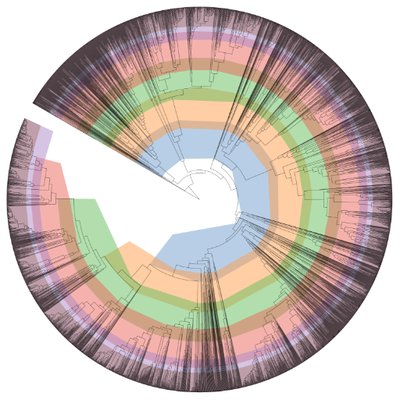
App: Insert Set of Genomes into SpeciesTree OR Insert Genome into Species Tree
Timing: 5-10 minutes
View Configure:
Genome Set: Use the annotated genome set from RAST that contains all your annotated bins from a sample.
Neighbor Public Genome Count: This is the number of additional genomes that will be added to the phylogenetic tree.
Copy Public Genomes to Workspace: Checking this box will add all the new genomes from the species tree into your data panel on the left. If you're stopping here you don't need to do this, but some analyses you would perform after this step might require you to save these genomes.
Output Tree: Name the tree that will be produced.
Output GenomeSet: Name the new GenomeSet (more important if you're saving all the public genomes)
Results: This app will generate a tree showing your assembled genomes highlighted in blue and additional genomes in white.


| Bin.004.fasta_assembly.RAST |
Q20 Did any of the genomes not have any close relatives indicated on the phylogenetic tree? Why do you think this happened?
Unfortunately I didn't find any Delftia in these samples, but if I did, I could run further analyses including metabolic modeling, multiple sequence alignments (MSA) of any genes of interest and further domain annotations.
The apps are inserted below, if you do find Delftia in your sample and are curious.



