Generated August 3, 2020
 Delftia is a genus of bacteria with a bunch of cool features! The best studied species, Delftia acidovorans can produce gold nanoparticles from gold ions in solution. This bacteria has been found living in biofolms with Cupriavidis metallidurans on gold nuggets(1). It is also found in soil, in sinks and in rhizospheres of different plants where it promotes their growth(3).
Delftia is a genus of bacteria with a bunch of cool features! The best studied species, Delftia acidovorans can produce gold nanoparticles from gold ions in solution. This bacteria has been found living in biofolms with Cupriavidis metallidurans on gold nuggets(1). It is also found in soil, in sinks and in rhizospheres of different plants where it promotes their growth(3).
Delftia acidovorans forms gold nuggets by producing a short nonribosomal peptide called delftibactin (1). The 16 genes responsible for the production of delftibactin are called the del cluster (delA-delP) (1). These genes were originally discovered in Delftia acidovorans SPH-1, but our research shows that the del cluster appears to be present across the genus (1).
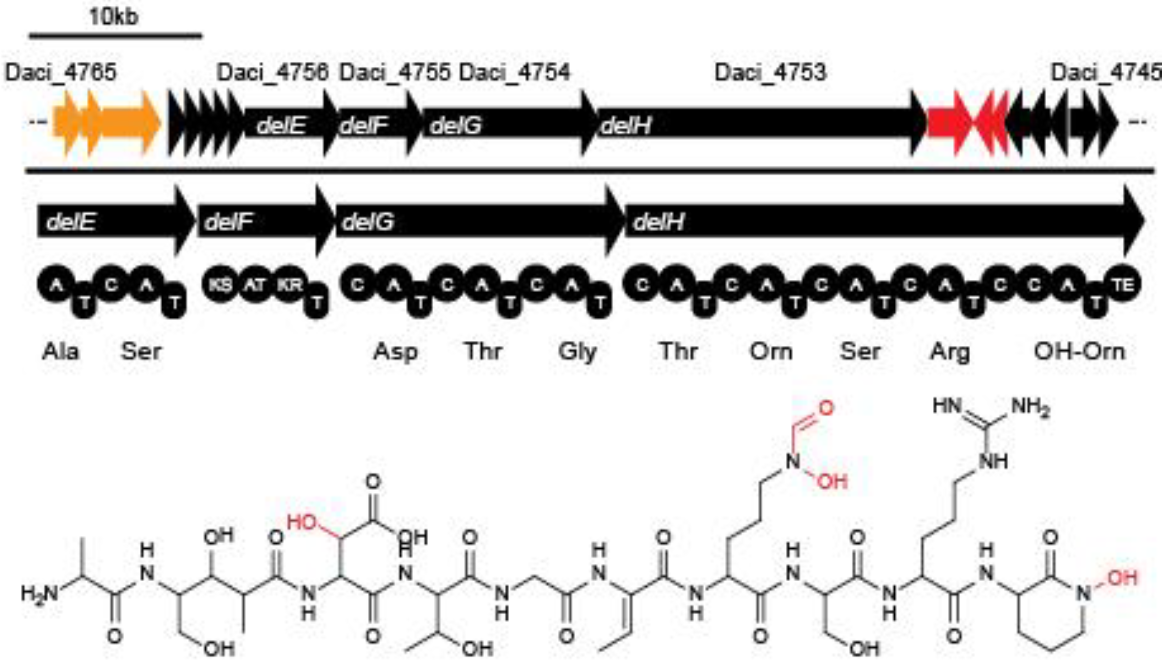
http://2013.igem.org/Team:Heidelberg/Project/Delftibactin
Proteobacteria
Betaproteobacteria
Burkholderiales
Comamonadaceae
Delftia
Our Workflow:
 Each step will be outlined before you reach it with a description of the apps, parameters and results.
Each step will be outlined before you reach it with a description of the apps, parameters and results.
We'll start with a number of raw reads, clean them up and do a little taxonomy to figure out what we might be able to assemble. Next, we'll assemble the reads using 4 different methods and pick out the best assembly. Then we'll take those reads and sort them by genome and assess the quality of those genomes. We'll select the high quality genomes to annotate and insert into phylogenetic trees to find relatives.
After completing this narrative, students will be able to:
Study Name:
Bio Sample:
Sample Name:
SRA:
Run ID:
Link to Sample:
The data I'm using comes from publicly available datasets available from NCBI's Sequence Read Archive (SRA).
The sample you need to upload should be a metagenomic sample listed as a WGS having paired reads. Your sample cannot be more than 20G bases or you'll need to import it through Globus (see this link for more information). You can also check out this really informative narrative for an in depth view of how to upload data from other sources: https://narrative.kbase.us/narrative/48493
App: Import SRA File as Reads from Web
Timing: 1-5 hours depending on the size of the file and queue time. (Its often helpful to run this in the morning so it uploads and then you can set up the assembly to run overnight or over the weekend.)
View Configure:
SRA URL: Use the first link from the "Reads Access" page and paste it into this block.
Reads Object Name: this will be what your sample is called once it's been uploaded into KBase. Make sure its something you'll be able to remember and follow the workflow with.
Sequencing Technology: Select the sequencing technology that was used to call the reads.
Single Genome: These reads are all metagenomes, so you don't need to select this box.
Results: Your reads should appear in the Data panel to the left. Before proceding, double check that they are a PairedEndLibrary. If they are a SingleEndLibrary, you'll need to select a different sample, because your assembly won't work with a SingleEndLibrary. In the results panel itself you'll see some stats from the reads including the number of reads, quality score mean and mean read length.

You've uploaded your data, great! Now you have to check the quality of the reads. First assess quality with FastQC. Then, if you need to, trim the reads and assess quality again to make sure the low quality reads have been removed from the sample.
Apps:
Timing: 5-20 minutes depending on queue and the number of reads
Assess Read Quality with FastQC View Configure:
Results: This app will give you a full report of the quality of your reads. The report will have two pages, for the forward and reverse reads in the PairedEndLibrary. Here we'll be focusing on the Per Base Sequence Quality, but the rest of the report offers a bunch of useful information about our library.
For a more in depth look at understanding your FastQC report, check out the manual here: https://dnacore.missouri.edu/PDF/FastQC_Manual.pdf
Trim Reads with Trimmommatic View Configure
Read library or set: Select the library you want to trim.
Parameters: This section covers different trimming parameters specific to removing adapters, croping the sequences, and the quality thresholds required to trim a read. In this workflow, I'm going to leave them all as defaults, but you can learn more about them in the App Info page and in the KBase App catalog.
Output library name: Make sure to specify that this library has been trimmed so you can tell the two libraries apart later.
Once you've trimmed the read library, reassess the quality of the PairedEndLibrary using FastQC.
Results: A FastQC Report that details the quality of the reads you've submitted and possibly trimmed libraries.


Before we assemble these libraries, it will be helpful to get an idea of what's present in our samples and at what abundance. KBase has two apps to do this, Kaiju and GOTTCHA2.
Apps:
Timing: 20 mins-2 hours depending on queue and number of reads you're running
1. Classify Taxonomy of Metagenomic Reads with Kaiju
This app translates reads into proteins and uses those sequences to identify what's present or possibly present in the sample.
View Configure:
Results: Your results will be a series of tables showing the breakdown of your sample beginning with the phyla and ending at species. The tail includes everything that is present below the low abundance filter.
2. Classify Taxonomy of Metagenomic Reads with GOTTCHA2
Unlike Kaiju, this app shows relative abundance based on unique nucleotide sequences from RefSeq.
View Configure:
Results: There are three ways you can view the results from GOTTCHA2. The first is as a table showing the classification of your reads, some statistics regarding their abundance and their relative abundance. The second is as a phylogenetic tree showing the relationships of the different taxa identified in your sample. The third layout is as a Krona plot, an interactive plot that displays relative abundance and phylogenetic relationships. Clicking on a phylum will zoom in to show the classes within it. How far you can zoom down depends on the sample and the unique sequences in it.
Run Kaiju twice. For the first run select the NCBI BLAST nr (no Euks) database. For the second run use the RefSeq (no Euks) database so you can compare the two results.

Alright, you've made it this far. This step takes the longest, so you may want to set it up to run overnight or over the weekend. This step will take our read libraries and line them up into longer sequences called contigs. Later we'll sort these contigs based on what genomes they came from. We'll be using three different apps to generate 4 different sets of contigs.
Apps:
Timing: hours to days (One of my assemblies below ran for almsot 4 days.)
1. Assemble Reads with MetaSPAdes
View Configure:
2. Assemble Reads with MEGAHIT (run 2x)
View Configure:
3. Assemble Reads with IDBA-UD
View Configure:
Results: Regardless of the assembly you use, you'll get the same result; a QUAST report. This report details the major features of your assembly and important statistics, such as the length of the longest read, the number of reads longer than 1,000,000 bases, the N50 and L50. In the next step we'll compare these statistics and receive a visual representation of the key parts of this report. For now, take note of which assembly contains the most base pairs.



Success! You've created 4 different assemblies from your metagenomic data. Now, you need to pick the best to sort out all those contigs into genomes. That step, sorting the contigs into different genome bins is called binning. Each bin will represent a single genome, but more on that later.
App: Compare Assembled Contig Distributions
Timing: 5-10 mins.
View Configure:
All you need to add here are your different assemblies from above to compare them and select the best to use moving forward.
The Results: A report showing the different statistics from each assembly. Some key features of this report include:
A good assembly will have:
A bad assembly will have:

Alright, you've picked the best assembly, now you'll sort all the contigs into bins that each represent a single genome. This step is called binning the contigs.
App: Bin Contigs using MaxBin2
Timing: 2+ hours depending on the number of contigs and bins present in your sample.
View Configure:
Assembly Object: Put your best assembly here.
Read Library: This is the library the contigs were generated from. If you needed to trim your read library, use the trimmed reads.
Probability Threshold: The confidence the alrogrithm must have for a contig to be placed within a bin. If a contig falls below this cutoff, then it will be left as unclassified. The default is 0.8.
Marker Set: MaxBin2 can bin both bacterial and archaeal genomes. In this case we're only looking at bacteria, so keep it set to the bacterial marker gene set.
Minimum contig length: Any contigs shorter than this will be ignored when binning. 1000 is the default, but above we set our contig minimum length at 2000 so we can increase this to 2000 or leave it as is, since we shouldn't have any contigs shorter than 2000 bases.
Results: The output from this app opens in a new section. The first panel lists the number of bins (and maximum number of genomes) and nucleotides included in all the contigs. The second tab offers some detail about the different bins including marker completenes, GC content, the number of contigs in each bin and their total length. To see information about the individual contigs in a bin, click the bulleted list icon for that bin or the graph beside it. However, these results tell you nothing about the quality of the bins, they could be highly contaminated or contain multiple copies of the same set of genes.

You should now have a bunch of different bins. Each bin represents a single genome (in theory). In this step we'll check these bins for their completeness, contamination and any duplicates using CheckM.
App: Assess Genome Quality with CheckM
Timing: Depends on the number of bins in your sample and the reference tree you pick
View Configure
Input Assembly, Genome or BinnedContigs: Add in your set of bins from the last step here.
Reference Tree: You can either select the full tree or reduced tree to compare your bins to. The full tree takes longer, but is recommended for a better understanding of what each bin represents. However, if you're tight on time the reduced tree is fine since we'll be generating a species tree to determine close relatives of our assemblies later.
Save all Plots: Save will allow you to download a .zip file of the resulting genome quality plots. Don't save will not.
Results: CheckM will give you two forms of the same report, a graphic version and a table. I think the table is easier to understand, so that's what I'll be covering here. The first column shows the bin name. They're all just numbered bins at this point, but you can rename them later if you want. The second column shows the lineage of the markers present in that bin. Some will be more specific than others, depending on the bin, its completeness and contamination. Number of genomes is the number of genomes used to create the marker set, and number of markers is the number of markers generated. These markers are unique and are expected to occur only once in the genome, replicates indicate contamination. The columns 0 through 5+ indicate the additional copies of these marker genes and are used to calculate contamination. Be aware that contamination is an underestimate in this app.
The last two columns indicate the completeness and contamination of your genome as percents. High quality genomes are over 90% complete with less than 5% contamination. However, since we're just looking to ID if Delftia is present, I'm using any genomes over 75% complete with less than 5% contamination. If an assembly falls outside this range, but looks promising, you can keep it, but be sure to note that it's a low quality assembly.
Write out a list of all the bins you want to keep, it will be useful in the next step.

If your assembly did not produce a high quality bin, select the most complete bin with the lowest level of contamination to use for the following steps. Make note of the completeness and contamination of this bin in your assignment questions.
Up above you checked the quality of all your bins and picked out all the ones that were over 75% complete and less than 5% contaminated. Now you're going to separate them from the contaminated and incomplete assemblies so you just have to work with them.
App: Extract Bins as Assemblies from BinnedContigs
Timing: Depends on the number of bins you're extracting, in general ~10 minutes or so
View Configure:
Binned Contigs: Select the binned contigs set you put into CheckM above. Once you add it the data will automatically fill into the lower Parameters section.
Bin Names Available for Extraction: There is a green plus on the right side of all your bins. Click it to select the ones you want to save as assemblies. They will appear in the lower table. Once you're done, double check that they're all there and that you got the right ones.
Assembly Name Suffix: Your bins will be renamed with this added. It should be a descriptive suffix so you can tell them apart, because any extracted bins will start with Bin###.fasta
AssemblySet Name: This will be the name of your assembly set that contains all your extracted bins. Again, it should be named something descriptive. For example, I named the first assembly set : Subsurface_gold_mine_extracted_bins.AssemblySet
If you have just one bin to extract your results will just be an assembly, because an AssemblySet needs to include 2 or more assemblies. This won't produce an error message.
Results: Your results will include a table of the different assemblies and a note that the job finished successfully.

You now have a set of mostly-whole, mostly-uncontaminated genomes from your sample. Now you'll use RAST to identify genes in these assemblies.
App: Annotate Multiple Microbial Assemblies (RAST)
Timing: Depends on the number of assemblies and their size. I'd estimate it takes about 10 mins an assembly.
View Configure:
Assemblies/AssemblySets: Here is where you add the AssemblySet you generated in the last step. You could add your assemblies individually, but it's easier to add them all as one set.
Domain and Genetic Code: Both should be set for bacteria, since D. acidovorans is a bacteria.
Call Buttons: By default most of these are checked. For something like a genome assembly, it's good to grab more features than we need in case we need them for a future study.
Results: Your results from RAST are fairly simple. You'll get objects for each assembly and one set of all assemblies annotated together. The summary will give you a short description of what was annotated in each genome and if the annotation was a success. Check through the summary to make sure none of the assemblies failed. Sometimes one will fail, but the app will still give you a success message. If it does fail, try annotating that assembly alone using the app: Annotate Microbial Assembly with different settings.

Now that you've annotated your assemblies, it's time to figure out what they are!
App: GTDB-Tk classify
Timing: about 30 minutes, depending on the number of assemblies and the queue time
View Configure:
Assembly Input: Add in your annotated assembly set from above.
Minimum Alignment Percent: This will filter out genomes with an insufficient percentage of AAs in the MSA generated by the app. The default is 10, if you want to increase the specificity all you need to do is increase this percentage. In my runs below I've kept the default as it is.
Results: The results of this app are across 4 tabs. The first tab is a table for bacteria and the second shows the same table for archaea. The first column indicates the bin and the second indicates the classification of that bin based on GenBank and RefSeq databases. The middle columns offer information about how this classification was determined. The right-most column is also important, since it will note any concerns about your genome. For example, if it contains high levels of contamination.
Lastly, I'll identify close relatives to my bins just to establish some addtional phylogenetic context for them.
App: Insert Set of Genomes into SpeciesTree OR Insert Genome into Species Tree
Timing: 5-10 minutes
View Configure:
Genome Set: Use the annotated genome set from RAST that contains all your annotated bins from a sample.
Neighbor Public Genome Count: This is the number of additional genomes that will be added to the phylogenetic tree.
Copy Public Genomes to Workspace: Checking this box will add all the new genomes from the species tree into your data panel on the left. If you're stopping here you don't need to do this, but some analyses you would perform after this step might require you to save these genomes.
Output Tree: Name the tree that will be produced.
Output GenomeSet: Name the new GenomeSet (more important if you're saving all the public genomes)
Results: This app will generate a tree showing your assembled genomes highlighted in blue and additional genomes in white.
